Resolución 458/2025
RESOL-2025-458-APN-PRES#SENASA
Ciudad de Buenos Aires, 25/06/2025
VISTO el Expediente N° EX-2025-57445616- -APN-DGTYA#SENASA; las Leyes Nros. 24.7676 y 27.233; el Decreto Reglamentario N° DECTO-2019-776-APN-PTE del 19 de noviembre de 2019, las Resoluciones Nros. 440 del 4 de agosto de 1998 y 350 del 30 de agosto de 1999 y sus modificatorias, ambas de la entonces SECRETARÍA DE AGRICULTURA, GANADERÍA, PESCA Y ALIMENTACIÓN; 801 del 10 de abril de 2015 de la SUPERINTENDENCIA DE RIESGOS DEL TRABAJO; 1.136 del 11 de agosto de 2000, 1.765 del 12 de octubre de 2000, 539 del 1 de julio de 2002, 371 del 1 de agosto de 2003, 822 del 10 de noviembre de 2011 y su modificatoria, 302 del 13 de junio de 2012, 389 del 24 de agosto de 2015, 671 del 23 de noviembre de 2016, RESOL-2017-660-APN-PRES#SENASA del 5 de octubre de 2017 y su modificatoria, RESOL-2018-829-APN-PRES#SENASA del 13 de noviembre de 2018, RESOL-2019-1684-APN-PRES#SENASA del 9 de diciembre de 2019, RESOL-2021-3-APN-PRES#SENASA del 5 de enero de 2021, RESOL-2023-1004-APN-PRES#SENASA del 12 de octubre de 2023 y su modificatoria y RESOL-2024-694-APN-PRES#SENASA del 26 de junio de 2024, todas del SERVICIO NACIONAL DE SANIDAD Y CALIDAD AGROALIMENTARIA, y
CONSIDERANDO:
Que mediante la Ley N° 27.233 se declara de interés nacional la sanidad de los animales y los vegetales, así como la prevención, el control y la erradicación de las enfermedades y de las plagas que afecten la producción silvoagropecuaria nacional, la flora y la fauna, la calidad de las materias primas producto de las actividades silvo-agrícolas, ganaderas y de la pesca, así como también la producción, inocuidad y calidad de los agroalimentos, los insumos agropecuarios específicos y el control de los residuos químicos y contaminantes químicos y microbiológicos en los alimentos y el comercio nacional e internacional de dichos productos y subproductos.
Que, asimismo, establece que el SERVICIO NACIONAL DE SANIDAD Y CALIDAD AGROALIMENTARIA (SENASA) es la autoridad de aplicación y el encargado de planificar, ejecutar y controlar el desarrollo de las acciones allí previstas.
Que a través del Decreto Reglamentario N° DECTO-2019-776-APN-PTE del 19 de noviembre de 2019 se aprueba la Reglamentación de la referida Ley N° 27.233.
Que por la Resolución N° 350 del 30 de agosto de 1999 de la entonces SECRETARÍA DE AGRICULTURA, GANADERÍA, PESCA Y ALIMENTACIÓN se aprueba el Manual de Procedimientos, Criterios y Alcances para el Registro de Productos Fitosanitarios en la REPÚBLICA ARGENTINA.
Que la citada norma establece los requisitos de composición, físicos, químicos, toxicológicos y ambientales a que están sujetas las sustancias activas químicas y bioquímicas de grado técnico.
Que la misma norma indica que la totalidad de la información toxicológica y ecotoxicológica presentada a los fines del registro de sustancias activas nuevas, debe estar avalada de acuerdo con la modalidad establecida por la autoridad competente.
Que la mentada Resolución N° 350/99 ha sufrido modificaciones a lo largo de los años para adaptarse a las nuevas realidades de las demandas del sector agrícola Argentino y a la necesidad de armonización con los avances científicos adoptados internacionalmente. Existe preocupación por reducir las plagas resistentes a los ingredientes activos disponibles actualmente en el mercado, a través de la disponibilidad de nuevas tecnologías que permitan un manejo adecuado de estas. Entre los objetivos de la actualización normativa se encuentran: aumentar la competencia en el mercado de productos fitosanitarios y similares, mejorar la seguridad de los aplicadores y estimular la investigación de nuevos productos fitosanitarios para su uso en el país.
Que la Resolución N° 389 del 24 de agosto de 2015 del SERVICIO NACIONAL DE SANIDAD Y CALIDAD AGROALIMENTARIA mantiene el “Registro de Profesionales Independientes Especializados en Toxicología y Ecotoxicología”, oportunamente creado por la Resolución N° 359 del 10 de septiembre de 1996 del ex-INSTITUTO ARGENTINO DE SANIDAD Y CALIDAD VEGETAL, y actualiza los requisitos a cumplimentar por parte de los profesionales interesados en inscribirse en el registro aludido.
Que resulta necesario actualizar los requisitos de la información toxicológica y ecotoxicológica para el registro de nuevas sustancias activas a ser registradas por equivalencia, según la normativa vigente.
Que el Artículo 1° de la Resolución N° RESOL-2023-1004-APN-PRES#SENASA del 12 de octubre de 2023 del citado Servicio Nacional establece el procedimiento de registro de bioinsumos para quienes se encuentren interesados en elaborar, importar, exportar, tener, fraccionar, distribuir y/o vender bioinsumos.
Que, por su parte, la Resolución N° 801 del 10 de abril de 2015 de la SUPERINTENDENCIA DE RIESGOS DEL TRABAJO aprueba la implementación del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA/GHS) en el ámbito laboral y, por ende, es necesario adoptar el SGA en las frases de advertencia y alerta utilizadas para comunicar el peligro en los marbetes y hojas de datos de seguridad de los productos fitosanitarios.
Que la adopción del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA/GHS) genera la necesidad de modificar la norma vigente en materia de etiquetado de productos fitosanitarios y de establecer los plazos razonables para su modificación y posterior adecuación de las etiquetas actualmente aprobadas.
Que por la Resolución N° RESOL-2024-694-APN-PRES#SENASA del 26 de junio de 2024 del aludido Servicio Nacional se establecen las condiciones y los requisitos para solicitar el reconocimiento de equivalencia de sustancias activas grado técnico, aprobadas por las autoridades competentes de determinados países o grupos de países.
Que resulta necesario abrogar la mencionada Resolución N° 694/24 a los efectos de actualizar y unificar los criterios y procedimientos relacionados con el reconocimiento de equivalencia de sustancias activas grado técnico, asegurando una mayor coherencia y eficacia en la normativa vigente.
Que corresponde a la Dirección de Agroquímicos y Biológicos, dependiente de la Dirección Nacional de Protección Vegetal del referido Servicio Nacional, en el ámbito de sus competencias, el establecimiento de los tiempos de carencia, los períodos de seguridad y los límites máximos de residuos de productos fitosanitarios en productos de origen vegetal y animal.
Que, por ende, es necesario actualizar los estándares exigidos por la Organización para la Cooperación y el Desarrollo Económico (OCDE).
Que resulta procedente armonizar los nuevos requisitos y procedimientos para todas las solicitudes, tanto futuras como las que se encuentran en trámite, a fin de igualar las exigencias para aquellas solicitudes previas a la fecha de vigencia de la presente.
Que la Dirección de Asuntos Jurídicos ha tomado la intervención que le compete.
Que el suscripto es competente para dictar el presente acto de conformidad con lo dispuesto por el Artículo 8°, inciso f), del Decreto N° 1.585 del 19 de diciembre de 1996 y sus modificatorios.
Por ello,
EL PRESIDENTE DEL SERVICIO NACIONAL DE SANIDAD Y CALIDAD AGROALIMENTARIA
RESUELVE:
ARTÍCULO 1°.- Aprobación. Se aprueba el nuevo Manual de Procedimientos, Criterios y Alcances para la autorización de establecimientos y/o de las personas humanas o jurídicas que intervengan en la cadena de elaboración en el mercado local, la importación y/o la exportación de productos fitosanitarios, de conformidad con lo dispuesto en la presente norma y los anexos que forman parte integrante de ella.
(Artículo sustituido por art. 1° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
ARTÍCULO 2°.- Autorización de personas humanas o jurídicas y/o establecimientos elaboradores. Todos los establecimientos elaboradores de productos fitosanitarios deben presentar una Declaración Jurada, conforme se detalla en el Anexo I que forma parte integrante de la presente resolución.
La presentación de dicha Declaración Jurada otorgará automáticamente a las personas humanas o jurídicas y a los establecimientos la autorización para iniciar sus actividades, quedando sujetos a la fiscalización posterior del SERVICIO NACIONAL DE SANIDAD Y CALIDAD AGROALIMENTARIA (SENASA).
(Artículo sustituido por art. 2° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
ARTÍCULO 3°.- Autorización para experimentación y ensayos a campo o invernadero con destino al control de plagas. La realización de actividades de experimentación y ensayos a campo o invernadero con destino al control de plagas agrícolas y silvícolas, que se encuentran en etapas tempranas de desarrollo debe ser autorizada por el SENASA, de conformidad con lo dispuesto en el Anexo II (IF-2025-68647702-APN-DNPV#SENASA), que forma parte integrante de la presente.
ARTÍCULO 4°.- Del Registro Nacional de Productos Fitosanitarios. Creación. Procedimientos de registro de productos fitosanitarios. Se crea el Registro Nacional de Productos Fitosanitarios que funcionará en el ámbito de la Dirección de Agroquímicos y Biológicos de la Dirección Nacional de Protección Vegetal del SENASA y se aprueba el procedimiento de registro de los productos fitosanitarios, conforme se detalla en el Anexo III, que forma parte integrante del presente acto administrativo.
Inciso a) A los fines del registro de un producto, el SENASA aceptará los resultados de los ensayos exigidos provenientes de laboratorios nacionales o extranjeros, siempre que se acredite fehacientemente el cumplimiento de Buenas Prácticas de Laboratorio (BPL o GLP) por parte de los referidos laboratorios. (Inciso sustituido por art. 3° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
Inciso b) Los productos fitosanitarios que hayan sido inscriptos con anterioridad a la creación del registro mencionado, serán incorporados de forma automática al mismo.
ARTÍCULO 5°.- De la validez de los registros de productos fitosanitarios. Los registros de productos fitosanitarios serán válidos indefinidamente, pudiendo ser cancelados eventualmente por el SENASA ante el incumplimiento de lo establecido en el presente manual, o por los motivos determinados en la normativa vigente o a solicitud de la persona humana o jurídica responsable del registro.
ARTÍCULO 6°.- Denegación o baja del Registro. El SENASA denegará o cancelará el registro de un producto fitosanitario objeto de la presente norma, si se determina técnica y científicamente que el producto representa un riesgo para la salud humana o si surgiera nueva información científica o epidemiológica que así lo demuestre.
En el supuesto caso de que un producto registrado en la REPÚBLICA ARGENTINA deje de estar autorizado en el país de origen, este será dado de baja automáticamente del Registro Nacional de Productos Fitosanitarios.
(Artículo sustituido por art. 4° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
ARTÍCULO 7°.- Importación de productos fitosanitarios. A los fines de la importación de productos fitosanitarios, se establece que:
Inciso a) Los productos fitosanitarios registrados y comercializados en los países con convergencia normativa listados en el Anexo IV de la presente resolución, podrán ser importados de conformidad con las siguientes condiciones/modalidades:
Apartado I) Aquellos que ya se comercializan para uso agrícola en la REPÚBLICA ARGENTINA ingresarán automáticamente mediante la presentación de una Declaración Jurada, acompañando la documentación indicada en el punto I “Información Administrativa” del Anexo V de la presente resolución, y un proyecto de marbete en idioma español, de acuerdo con la normativa vigente de etiquetado de productos fitosanitarios, considerando los usos autorizados en el país para el producto fitosanitario de referencia.
Apartado II) Aquellos que aún NO tienen antecedentes de registro y uso agrícola en la REPÚBLICA ARGENTINA deberán presentar una Declaración Jurada en la que conste que el producto no representa un riesgo para la salud humana, animal ni para el ambiente en el Territorio Nacional.
Subapartado i) La presentación de la Declaración Jurada otorgará al interesado un registro transitorio para el uso o la comercialización del producto fitosanitario, por el plazo máximo de DOS (2) años. Durante la vigencia del registro transitorio, el solicitante deberá realizar los ensayos de eficacia agronómica y determinación de residuos conforme a lo establecido en la presente, con el alcance y la periodicidad que indique el SENASA.
Subapartado ii) Dichos estudios deberán acreditar que el uso del producto, bajo las condiciones propuestas de aplicación, no implica riesgos inaceptables para la salud humana, animal ni para el ambiente, y que los residuos detectados en los productos de cosecha se encuentran dentro de los límites máximos permitidos para su consumo. El análisis de riesgo que sustente la solicitud debe ser realizado mediante procedimientos internacionalmente aceptados en países de alta vigilancia epidemiológica.
Subapartado iii) Durante el plazo en el que el SENASA no disponga de información suficiente desarrollada en la REPÚBLICA ARGENTINA para determinar el límite máximo de residuo (LMR) en cada cultivo o alimento tratado, se tomará como referencia el establecido por el país en el que se registró el producto. En caso de que el cultivo o el alimento tratado no tenga un LMR establecido en el país de convergencia normativa, se tomará como referencia el límite de cuantificación analítica (LOQ).
Subapartado iv) Los cultivos tratados con dicho producto fitosanitario solo podrán ser comercializados una vez que se hayan evaluado los ensayos correspondientes. Verificada la conformidad de los datos, el SENASA otorgará el Registro con carácter definitivo.
Subapartado v) Quedan excluidos del alcance del presente apartado los productos fitosanitarios de naturaleza biológica, los Organismos Modificados Genéticamente (OGM) sin antecedentes en el país y las sustancias derivadas u obtenidas con uso de OGM sin antecedentes en el país, productos desarrollados con nuevas técnicas de mejoramiento (NBT) y nuevas tecnologías con escasos o nulos antecedentes a nivel mundial.
Asimismo, se considerarán aquellos OGM que se encuentren aprobados por la Comisión Nacional Asesora de Biotecnología Agropecuaria (CONABIA), conforme al procedimiento establecido en la normativa vigente.
Subapartado vi) Si el producto deja de estar autorizado en el país donde fue registrado, quedará automáticamente excluido del alcance del presente artículo y será incluido en el procedimiento establecido en el Anexo VII - REEVALUACION DE PRODUCTOS FITOSANITARIOS REGISTRADOS, que forma parte integrante de la presente resolución.
Apartado III) En ambos supuestos, si el documento oficial de registro y comercialización emitido por la Autoridad Competente que integre el listado de países mencionado en el referido Anexo IV, no contempla la declaración de pureza e impurezas para sustancias activas y la declaración de composición en los formulados (información confidencial), los interesados deberán declarar dicha información, la que será reservada de acuerdo con la Ley N° 24.766.
Inciso b) Los productos fitosanitarios que no cuenten con registro y autorización de comercialización para uso agrícola y provengan de países que NO se encuentran listados en el citado Anexo IV, deberán realizar el registro completo, conforme se detalla en la presente norma.
Inciso c) Todas las solicitudes serán tramitadas a través de la Plataforma de Trámites a Distancia (TAD) o la que en el futuro la reemplace.
Inciso d) En todos los casos mencionados en los incisos precedentes de este artículo, el SENASA se reserva la facultad de llevar a cabo fiscalizaciones posteriores.
Inciso e) A los fines de la aplicación de la normativa vigente, serán oficialmente válidos todos aquellos modelos de Formularios de Inscripción, Constancias, Certificados y Notificaciones y Guías de Procedimientos disponibles o expedidos a través de la plataforma de gestión indicada por el SENASA a tales efectos.
(Artículo sustituido por art. 5° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
ARTÍCULO 8°.- Importación de muestras de productos fitosanitarios en etapas tempranas de desarrollo. En aquellos casos en los que se desee importar muestras de productos fitosanitarios en etapas tempranas de desarrollo, se deberá presentar un aviso, con carácter de Declaración Jurada, a través de la Plataforma SIG-Trámites, o la que en el futuro la reemplace, conforme se detalla en el Anexo VI que forma parte integrante de la presente medida. El producto solo podrá ser utilizado en el predio de experimentación en que se realizará la prueba.
(Artículo sustituido por art. 6° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
ARTÍCULO 9°.- De los productos fitosanitarios destinados a la exportación. En el caso de los productos destinados exclusivamente a la exportación, estos deberán cumplir con las reglamentaciones vigentes exigidas por el país de destino. A tal fin, el SENASA deberá otorgar las certificaciones correspondientes si le fueran solicitadas por los exportadores.
ARTÍCULO 10.- Extracción de muestras. El SENASA podrá efectuar la extracción de muestras de productos fitosanitarios para su posterior estudio en laboratorio, a fin de constatar que la información declarada se corresponda con lo informado por el solicitante.
ARTÍCULO 11.- Adecuación toxicológica. Las empresas que posean productos inscriptos actualmente en el Registro Nacional de Terapéutica Vegetal o en el Registro Nacional de Productos Fitosanitarios que se crea en la presente norma, cuentan con un plazo máximo de TRES (3) años para adecuarse a la implementación del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA).
(Artículo sustituido por art. 7° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
ARTÍCULO 12.- Etiquetado. Plazos. Se fija un plazo de NOVENTA (90) días, contados a partir de la vigencia de la presente, para establecer las nuevas condiciones de etiquetado de los productos fitosanitarios con la incorporación de la clasificación de los productos mediante la versión del Manual de las Naciones Unidas del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA), adoptado en la presente, y los plazos de adecuación de las etiquetas actualmente aprobadas con otra clasificación.
ARTÍCULO 13.- Reevaluación de productos fitosanitarios registrados. El SENASA puede determinar una reevaluación del uso de un producto fitosanitario de acuerdo con lo dispuesto en el Anexo VII (IF-2025-68651146-APN-DNPV#SENASA), que forma parte integrante de la presente norma.
ARTÍCULO 14.- Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA). A fin de garantizar la seguridad en el manejo de sustancias químicas y proteger la salud de las personas, resulta indispensable establecer criterios uniformes para la identificación de los peligros asociados. En este sentido, la clasificación de los peligros para la salud deberá realizarse conforme a lo dispuesto en la Versión ST/SG/AC.10/30/Rev.9 del Manual de las Naciones Unidas del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA).
Asimismo, podrá utilizarse como guía complementaria lo determinado en el Anexo VIII, que forma parte integrante del presente acto, para la implementación práctica de la clasificación y comunicación de los peligros para la salud.
ARTÍCULO 14 bis.- Guía de Procedimientos para la Gestión de Productos Fitosanitarios. Aprobación. Se aprueba la “Guía de Procedimientos para la Gestión de Productos Fitosanitarios” de la Dirección Nacional de Protección Vegetal del SENASA, la cual estará disponible en el sitio web oficial del Organismo.
(Artículo incorporado por art. 8° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
ARTÍCULO 15.- Se faculta a la Dirección Nacional de Protección Vegetal del SENASA a tomar las medidas y los recaudos necesarios para extender el alcance de los requisitos y procedimientos de la presente norma a las solicitudes que se encuentran en trámite a la fecha de vigencia de esta resolución.
ARTÍCULO 16.- Incumplimiento. Sanciones. El incumplimiento o las transgresiones a la presente norma serán pasibles de las sanciones establecidas en el Capítulo V de la Ley N° 27.233 y su Decreto Reglamentario N° DECTO-2019-776-APN-PTE del 19 de noviembre de 2019, sin perjuicio de las acciones preventivas que pudieran adoptarse en virtud de lo dispuesto en la Resolución N° 38 del 3 de febrero de 2012 del entonces MINISTERIO DE AGRICULTURA, GANADERÍA Y PESCA y su modificatoria, o la que en el futuro la reemplace.
ARTÍCULO 17.- Anexos. Aprobación. Se aprueban los siguientes anexos, los cuales forman parte integrante de la presente resolución:
Inciso a) Anexo I “AUTORIZACIÓN DE PERSONAS HUMANAS O JURÍDICAS Y ESTABLECIMIENTOS ELABORADORES” (IF-2025-120999108-APN-DNPV#SENASA).
Inciso b) Anexo II “AUTORIZACIÓN DE PREDIOS DE EXPERIMENTACIÓN DE PRODUCTOS FITOSANITARIOS EN ETAPAS TEMPRANAS DE DESARROLLO” (IF-2025-120999543-APN-DNPV#SENASA).
Inciso c) Anexo III “REGISTRO DE PRODUCTOS FITOSANITARIOS” (IF-2025-121541245-APN-DNPV#SENASA).
Inciso d) Anexo IV “LISTADO DE PAÍSES/GRUPO DE PAÍSES CON CONVERGENCIA NORMATIVA” (IF-2025-121541330-APN-DNPV#SENASA).
Inciso e) Anexo V “AVISO DE IMPORTACIÓN DE PRODUCTOS FITOSANITARIOS” (IF-2025-68648196-APN-DNPV#SENASA).
Inciso f) Anexo VI “AVISO DE IMPORTACIÓN DE MUESTRAS DE PRODUCTOS FITOSANITARIOS EN ETAPAS TEMPRANAS DE DESARROLLO” (IF-2025-121006670-APN-DNPV#SENASA).
Inciso g) Anexo VII “REEVALUACIÓN DE PRODUCTOS FITOSANITARIOS REGISTRADOS” (IF-2025-68651146-APN-DNPV#SENASA).
Inciso h) Anexo VIII “CLASIFICACIÓN, COMUNICACIÓN DE LOS PELIGROS PARA LA SALUD, Y PROTOCOLO DE ENSAYOS DE EFICACIA AGRONÓMICA, FITOTOXICIDAD Y RESIDUOS” (IF-2025-121331444-APN-DNPV#SENASA).
(Artículo sustituido por art. 9° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
ARTÍCULO 18.- Abrogaciones. Se abrogan las Resoluciones Nros. 440 del 4 de agosto de 1998 y 350 del 30 de agosto de 1999, ambas de la entonces SECRETARÍA DE AGRICULTURA, GANADERÍA, PESCA Y ALIMENTACIÓN, 1.136 del 11 de agosto de 2000, 1.765 del 12 de octubre de 2000, 539 del 1 de julio de 2002, 371 del 1 de agosto de 2003, 822 del 10 de noviembre de 2011, 302 del 13 de junio de 2012, RESOL-2017-660-APN-PRES#SENASA del 5 de octubre de 2017, RESOL-2018-829-APN-PRES#SENASA del 13 de noviembre de 2018, RESOL-2021-3-APN-PRES#SENASA del 5 de enero de 2021, RESOL-2023-1004-APN-PRES#SENASA del 12 de octubre de 2023 y RESOL-2024-694-APN-PRES#SENASA del 26 de junio de 2024, todas del SERVICIO NACIONAL DE SANIDAD Y CALIDAD AGROALIMENTARIA.
ARTÍCULO 19.- Incorporación. Se incorpora la presente resolución al Libro Tercero, Parte Cuarta, Título II, Capítulo II, Sección 1a, del Índice Temático del Digesto Normativo del SERVICIO NACIONAL DE SANIDAD Y CALIDAD AGROALIMENTARIA, aprobado por Resolución N° 401 del 14 de junio de 2010 y su complementaria N° 913 del 22 de diciembre de 2010, ambas del citado Servicio Nacional.
ARTÍCULO 20.- Vigencia. La presente resolución entra en vigencia a partir del 5 de enero de 2026.
(Artículo sustituido por art. 10 de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
ARTÍCULO 21.- Comuníquese, publíquese, dese a la DIRECCIÓN NACIONAL DEL REGISTRO OFICIAL y archívese.
Pablo Cortese
NOTA: El/los Anexo/s que integra/n este(a) Resolución se publican en la edición web del BORA -www.boletinoficial.gob.ar-
e. 26/06/2025 N° 44548/25 v. 26/06/2025
(Nota Infoleg: Los anexos referenciados en la presente norma han sido extraídos de la edición web de Boletín Oficial)

AUTORIZACIÓN DE PERSONAS HUMANAS O JURÍDICAS Y ESTABLECIMIENTOS ELABORADORES
Todas las personas humanas o jurídicas que comercialicen, importen, para uso directo y/o los establecimientos que sinteticen o formulen productos fitosanitarios deberán solicitar una autorización con carácter de Declaración Jurada a través de la plataforma Trámites a Distancia (TAD), o la que en el futuro la reemplace.
Dicha autorización deberá contener los datos que a continuación se detallan:
Autorización de Persona Humana:
a) Nombre y apellido.
b) C.U.I.T. / C.U.I.L.
c) Teléfono.
d) Correo electrónico.
e) Nombre del Representante Legal y su correo electrónico.
Autorización de Personas Jurídicas:
a) Razón Social.
b) C.U.I.T. / C.U.I.L.
c) Dirección, teléfono y correo electrónico.
d) Nombre del Representante Legal y su correo electrónico.
Autorización de Establecimiento Elaborador:
a) Nombre y dirección de la compañía, persona humana o jurídica.
b) Nombre, dirección del establecimiento elaborador para el que se solicita y georreferenciación en coordenadas decimales (Decimal Degrees —DD—).
c) Para establecimientos situados en la República Argentina C.U.I.T. / C.U.I.L.
d) Teléfono y correo electrónico.
Cualquier modificación de las condiciones en las que fue otorgada la autorización, deberá ser comunicada a la Autoridad de Aplicación a través de sus sistemas informáticos.
La baja de la autorización podrá ser solicitada por la firma responsable del establecimiento o también producto de una sanción aplicada por el SENASA, como conclusión de un trámite sumarial del cual surge clara y fundadamente la gravedad del incumplimiento a la normativa vigente.


AUTORIZACIÓN DE PREDIOS DE EXPERIMENTACIÓN DE PRODUCTOS FITOSANITARIOS EN ETAPAS TEMPRANAS DE DESARROLLO
REQUISITOS GENERALES
Las personas humanas o jurídicas interesadas en realizar experimentaciones a campo y/o en invernadero que impliquen la liberación al agroecosistema de productos fitosanitarios en etapas tempranas de desarrollo deberán solicitar una autorización con carácter de declaración jurada a través de la plataforma SIG-Trámites, o la que en el futuro la reemplace.
La autorización y las obligaciones del solicitante para la liberación de productos fitosanitarios en etapas tempranas comprende todas las etapas involucradas en el manejo bioseguro de los materiales en evaluación, desde el ingreso al país, el almacenamiento, el proceso de experimentación, la cosecha y la disposición final del material vegetal interviniente y los posibles remanentes de las sustancias evaluadas. Asimismo, comprende el monitoreo ambiental posterior del sitio de la liberación utilizado, por el período que se determine en la respectiva autorización.
Las personas humanas o jurídicas responsables del predio experimental tienen la obligación de facilitar el acceso a las instalaciones del predio y a los registros documentales, a los evaluadores del SENASA.
El SENASA otorgará dicha autorización en un plazo no mayor de TREINTA (30) días hábiles.
INFORMACIÓN ADMINISTRATIVA.
Solicitud de inscripción del predio de experimentación de productos fitosanitarios en etapas tempranas de desarrollo, con carácter de declaración jurada, firmada por el Propietario, Apoderado o Representante Legal y el Responsable Técnico con incumbencia profesional en la materia.
En dicha solicitud se deberá completar la siguiente información:
a) Nombre y Apellido/Razón Social.
b) C.U.I.T. / C.U.I.L.
c) Dirección del establecimiento y Georreferenciación.
d) Teléfono.
e) Correo electrónico.
f) Título profesional habilitante de los profesionales calificados involucrados directamente en actividades de investigación y experimentación con productos fitosanitarios.
CUERPO TÉCNICO
DESCRIPCIÓN DE LA ENTIDAD DE INVESTIGACIÓN:
Organigrama de la entidad, currículum vitae resumido y nota de responsabilidad técnica del Director de la unidad experimental, y de los profesionales calificados involucrados directamente en actividades de investigación y experimentación con productos fitosanitarios, desde la planificación hasta la emisión de informes técnicos.
DESCRIPCIÓN DEL ESTABLECIMIENTO:
- Croquis de la estación experimental con ubicación y memorial descriptivo que informe el área total y área de experimentación e investigación, estado de conservación de suelos, ubicación de cuerpos de agua y áreas aledañas.
-Croquis de acceso a la estación experimental, con las Coordenadas Geodésicas (Datum WGS84, expresados en grados y SEIS (6) decimales de grado), declarando las coordenadas geográficas del acceso al establecimiento y de los vértices que contengan la totalidad de la superficie del predio experimental.
-Croquis de ubicación de instalaciones/sectores: oficinas, laboratorios, invernáculos, zona de investigación/desarrollo de campo, depósitos. Las instalaciones deben contar con alambrado perimetral.
- Inventario de máquinas, equipos agrícolas, instalaciones físicas, medios técnicos y materiales. Las instalaciones deben contar con estación meteorológica, acceso a internet, telefonía celular y grupo electrógeno.
MANUAL DE PROCEDIMIENTOS:
-Flujo de las actividades a desarrollar, debiendo constar procedimientos de recibo, almacenamiento, transporte, manipuleo, utilización y disposición final de las muestras de sustancias experimentales.
- Detalle de la relación entre la zona geográfica elegida y el objetivo de la investigación.
- Indicación de los sistemas de registro del ingreso, flujo y destino de las muestras en proceso de experimentación temprana.
- Caracterización y tratamiento de los residuos especiales aprobados por la Autoridad Ambiental Competente a nivel municipal o provincial.
- Caracterización del tratamiento de envases vacíos y material descartable aprobado por la Autoridad Ambiental Competente a nivel municipal o provincial.
- Seguridad operativa en el manejo de sustancias químicas: debe indicar las políticas, actividades y normas internas de la empresa o institución en esta materia (equipos de seguridad, ropa protectora, control de efluentes, disposición final de muestras, destrucción de cultivos y otros).
- Plan de Control y Monitoreo Ambiental de los recursos suelo/agua/aire, y medidas de aislamiento y bioseguridad.
- Caracterización y tratamiento de los residuos de los materiales vegetales tratados.
- Plan de contingencias para el manejo de accidentes con sustancias químicas e incendios, elaborado por un profesional competente.
SISTEMA DE REGISTRO:
- Libro de estudio y de campo.
- Registro de ingreso, almacenamiento y disposición final de las muestras experimentales.
- Registro de aplicación, condiciones de aplicación y destrucción de material tratado.
- Registro de mantenimiento de equipos.
- Registro de capacitaciones del personal involucrado.
- Registro de almacenamiento y disposición final de los residuos generados en el predio experimental.
ARCHIVO:
- Archivo de plan de estudio, libros de campo y otros registros de la entidad.

REGISTRO DE PRODUCTOS FITOSANITARIOS
Los productos fitosanitarios que se elaboren o formulen en la REPÚBLICA ARGENTINA o en otros países y no cuenten con el registro y la aprobación de uso agrícola por Autoridades Competentes del listado de países o grupos de países que se detallan en el Anexo IV deberán estar aprobados y registrados en el Registro Nacional de Productos Fitosanitarios, dependiente del SERVICIO NACIONAL DE SANIDAD Y CALIDAD AGROALIMENTARIA (SENASA) cumpliendo con lo establecido en el presente Anexo.
Los interesados que deseen registrar los productos deberán presentar un formulario de solicitud de registro dependiendo del tipo de producto a registrar, a través de la plataforma SIG-Trámites o la que en el futuro la reemplace, y la documentación correspondiente según el tipo de producto.
El SENASA procederá a evaluar la información presentada y expedirse en un plazo no mayor a CIENTO OCHENTA (180) días hábiles.
A los efectos de la presente norma los productos a registrar estarán encuadrados en DOS (2) categorías dependiendo del tipo de producto a registrar:
SUSTANCIAS ACTIVAS: se entiende por sustancia activa al componente principal de un producto fitosanitario que tiene la capacidad de ejercer un efecto biológico. Esta puede ser grado técnico nueva o equivalente.
SUSTANCIAS ACTIVAS NUEVAS: se considera sustancia activa grado técnico nueva a la que no ha sido registrada en el país.
En los casos en que se desee registrar este tipo de sustancias se deberá proceder al registro completo detallado en los apartados I a VI.
SUSTANCIAS ACTIVAS EQUIVALENTES: se considera sustancia activa equivalente cuando su equivalencia ha sido demostrada respecto de otras ya registradas en el país.
Las sustancias activas de diferentes fabricantes o de diferentes procesos de síntesis del mismo fabricante son equivalentes si:
- La sustancia activa tiene una concentración declarada mayor o igual a la de la referencia.
- El máximo nivel de impurezas no relevantes no se incrementa por más del CINCUENTA POR CIENTO (50 %) (relativo al máximo nivel en la referencia), o el nivel absoluto no se incrementa en más del CERO COMA TRES POR CIENTO PESO EN PESO (0,3 %p/p), considerando el que represente el mayor nivel de incremento.
- No se presentan nuevas impurezas mayores a CERO COMA UNO POR CIENTO PESO EN PESO (0,1 %p/p).
- No se encuentran impurezas relevantes por encima de los límites establecidos en la Resolución N° 481 del 27 de octubre de 2014 del citado Servicio Nacional y sus modificatorias, o en las especificaciones de la Organización de las Naciones Unidas para la Alimentación y la Agricultura (FAO).
En los casos en que se desee registrar este tipo de sustancias activas equivalentes, se deberán cumplimentar los apartados detallados en los puntos I a III.
PRODUCTO FORMULADO: se entiende por producto formulado a la mezcla de sustancias activas y otras sustancias, que cumple la función de proteger a las plantas contra plagas, enfermedades y malezas.
Los coadyuvantes incluidos como componentes de una formulación no serán pasibles de registro, pero deberán usarse para la elaboración o fabricación de productos fitosanitarios aquellos que no se encuentren alcanzados en la Resolución N° RESOL-2019-32-APN-PRES#SENASA del 17 de enero de 2019 del mencionado Servicio Nacional, o la que en un futuro la reemplace.
A fin de realizar la inscripción de fitosanitarios al registro, los requerimientos para las diferentes categorías son los siguientes:
A - SUSTANCIAS ACTIVAS
I - INFORMACIÓN ADMINISTRATIVA
Formulario de solicitud de registro, dependiendo del tipo de producto a registrar, firmado por el Apoderado o Representante Legal y el Responsable Técnico con incumbencia profesional en la materia.
Patrones: en caso de que el SENASA así lo requiera para control, la empresa registrante deberá aportar los patrones analíticos de la sustancia activa e impurezas, en las condiciones que establezca la Dirección General de Laboratorios y Control Técnico (DGLCyT) del SENASA.
Hoja de Datos de Seguridad: de acuerdo con la versión del Manual de las Naciones Unidas del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA), adoptada en la presente norma.
Informes técnicos de análisis de riesgo toxicológico y ecotoxicológico de nuevos principios activos de uso agrícola, firmados por los equipos interdisciplinarios de profesionales independientes especializados en toxicología y ecotoxicología del SENASA.
Si se desea registrar una sustancia activa grado técnico equivalente que se produzca en el país o que se importe desde países no incluidos en el Anexo IV, para acreditar dicha equivalencia se deberá presentar ante el SENASA el formulario de solicitud de registro, junto con toda la documentación técnica y administrativa requerida.
II - GENERALIDADES
Protocolos y estudios de Productos Fitosanitarios
Los estudios biológicos para la producción de datos toxicológicos y ecotoxicológicos y los laboratorios que realicen propiedades físicas y químicas, y determinaciones analíticas de residuos de principios activos en matrices vegetales y ambientales con fines de registro, revalidación, revaluación o monitoreo de productos fitosanitarios, deberán incluir la documentación respaldatoria correspondiente al monitoreo de Buenas Prácticas de Laboratorio (BPL), desarrolladas por la Organización para la Cooperación y el Desarrollo Económico (OCDE), emitida por un Organismo de Acreditación de reconocimiento internacional. Asimismo, los ensayos de residuos de principios activos deberán encuadrarse en la Directiva OCDE sobre aplicación de las BPL para los estudios de campo.
Los protocolos de los estudios biológicos para la producción de datos toxicológicos y de propiedades físicas y químicas y las determinaciones analíticas de residuos de principios activos deben realizarse mediante los protocolos correspondientes a los organismos y cuerpos normativos que protocolizan ensayos y procedimientos de laboratorio para la obtención de datos con fines de registro, a saber:
APVMA: AUSTRALIAN PESTICIDES AND VETERINARY MEDICINES AUTHORITY
• CIPAC: COLLABORATIVE INTERNATIONAL PESTICIDES ANALYTICAL COUNCIL
• EEC/EU: EUROPEAN ECONOMIC COMMUNITY/EUROPEAN UNION
• EFSA: EUROPEAN FOOD SAFETY AUTHORITY
• EPA: UNITED STATES ENVIRONMENTAL PROTECTION AGENCY
• EURACHEM: EUROPEAN COLLABORATION ON CHEMICAL MEASUREMENT
• FDA: UNITED STATES FOOD AND DRUG ADMINISTRATION
• FFDCA: FEDERAL FOOD DRUG AND COSMETIC ACT
• FIFRA: FEDERAL INSECTICIDE, FUNGICIDE AND RODENTICIDE ACT
• ISO: INTERNATIONAL STANDARD ORGANIZATION
• IUPAC: INTERNATIONAL UNION OF PURE AND APPLIED CHEMISTRY
• OECD: ORGANIZATION FOR ECONOMIC COOPERATION AND DEVELOPMENT
• OMS: WORLD HEALTH ORGANIZATION
• OPPTS: OFFICE OF PREVENTION, PESTICIDES AND TOXIC SUBSTANCES
Otros protocolos provenientes de organismos o cuerpos normativos diferentes a los que figuran en el presente Manual deberán ser consultados previamente al SENASA.
Clasificación toxicológica
Se adoptará como clasificación toxicológica la del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA/GHS), utilizando para tal fin la información sobre toxicología en mamíferos y no mamíferos de la sustancia activa o del producto formulado, según corresponda.
A los fines de comunicar el peligro en los marbetes de productos fitosanitarios y las hojas de datos de seguridad de estos, se deberá proceder de acuerdo con los criterios definidos en la Versión ST/SG/AC.10/30/Rev.9 del Manual de las Naciones Unidas del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA), y en función de la normativa de etiquetado de productos fitosanitarios vigente en la REPÚBLICA ARGENTINA, versión revisable al efecto del presente manual cada CINCO (5) años.
Accesibilidad de la información de registro
La información confidencial y los datos de pruebas serán provistos por el registrante, distinguiendo claramente entre qué es información confidencial y qué son datos de pruebas, considerando: Información confidencial: la información correspondiente a la identidad, composición, proceso de síntesis, proceso de formulación y otros secretos industriales y comerciales.
Datos de pruebas: datos de ensayos toxicológicos, ecotoxicológicos, de residuos y propiedades físicas y químicas.
Respecto a la información confidencial o a los datos no divulgados requeridos para su evaluación, el SENASA tomará las medidas necesarias para su salvaguarda contra todo uso comercial deshonesto y evitar que dicha información sea revelada o transferida. A tales efectos, el SENASA se encargará de proveer las instalaciones y los medios necesarios para garantizar la seguridad y una adecuada gestión de la información.
A tal fin, la información confidencial y los datos no publicados recibidos serán resguardados informáticamente en los sistemas que disponga especialmente el SENASA, y será proporcionada a los técnicos evaluadores a tal fin, no pudiendo estos efectuar copias ni totales ni parciales, ni compartir dicha información o darla a conocer a terceros.
Los técnicos evaluadores (funcionarios internos o auxiliares) dejarán constancia escrita de que aceptan las condiciones de confidencialidad en que deberán manejar el material informativo que se les provea, bajo declaración jurada.
Concluida la evaluación, la información confidencial y los datos no publicados serán resguardados siguiendo las medidas de seguridad dispuestas. El SENASA podrá disponer su entrega al administrado para que éste los tenga en guarda. En este último caso, la información confidencial y los datos no publicados deben estar disponibles en el caso que el SENASA lo solicite.
La información confidencial de las sustancias activas grado técnico consideradas de referencia para la determinación de equivalencia quedan en guarda del SENASA y no están alcanzadas por lo dispuesto en el párrafo previo respecto de su entrega al administrado.
Cuando los datos de ensayos y pruebas sobre seguridad y eficacia, resguardados en el Organismo, hubieran caído en el dominio público en cualquier país por la publicación de cualquiera de los datos protegidos, la presentación de todos o parte de estos datos en medios científicos o académicos o por cualquier otro medio de publicación, entonces estos dejarán de ser archivados mediante las medidas de seguridad dispuestas por el SENASA.
Los expedientes técnicos de registro son reservados de acuerdo con el marco previsto en el Artículo 38 del Decreto N° 1.759 del 3 de abril de 1972, sus modificatorios y complementarios. Su vista queda reservada al personal y los auxiliares de este Servicio Nacional afectados al procedimiento de registro, y a las personas fehacientemente autorizadas por el administrado titular del registro.
El SENASA utilizará la información suministrada del modo antes descrito a los efectos de los registros de productos fitosanitarios, observando el marco legal que impone la Ley N° 24.766 y normas complementarias.
Quedan expresamente exceptuados de la confidencialidad:
a) Nombre, contenido y origen de principios activos en productos formulados, establecimientos elaboradores y empresas registrantes.
b) Métodos y recomendaciones de transporte, almacenaje, tratamientos de incendio y otros riesgos.
c) Medios de disposición de envases.
d) Procedimientos de descontaminación.
e) Primeros auxilios y ayuda médica en caso de daño a las personas.
f) Método de análisis de residuos.
g) Método de análisis de las impurezas de importancia toxicológica o ecotoxicológica (denominadas de declaración obligatoria).
h) La información contenida en la Hoja de Datos de Seguridad.
i) Toda información que haya caído en el dominio público.
Quienes requieran la información referida en los incisos f) y g), deberán hacerlo mediante nota, expresando el motivo del requerimiento, la que será registrada y archivada.
El personal afectado a los procedimientos de registro de productos fitosanitarios se encuentra comprendido en los mandatos de los Artículos 3°, 12 y 13 de la Ley N° 24.766, por lo que deberá abstenerse de usar y de revelar sin causa justificada o sin consentimiento del registrante la información en cuestión, bajo apercibimiento de las sanciones que la misma norma prevé.
III - INFORMACIÓN CONFIDENCIAL (tanto para sustancias activas nuevas, como equivalentes)
a.- Certificado de Origen, el cual debe ser original y emitido por el establecimiento elaborador.
Debe incluir:
Identificación de la sustancia activa.
Contenido mínimo declarado porcentaje PESO EN PESO (p/p).
Nombre y localización del establecimiento elaborador.
Clave o número con el que la Autoridad Competente del país de origen identifica al establecimiento.
Nombre y dirección de la empresa que registrará el producto ante SENASA. En caso de que no figure esta información se deberá remitir una nota emitida por el establecimiento elaborador donde conste la autorización para realizar el registro del producto en el país.
b - Declaración de composición cuali-cuantitativa firmada por el establecimiento elaborador o por la empresa registrante con carácter de declaración jurada.
Debe incluir:
Nombre químico según criterios internacionales [Unión Internacional de Química Pura y Aplicada (IUPAC) o Chemical Abstract Service (CAS)] y número CAS (cuando esté disponible) de la sustancia activa y sus impurezas asociadas.
Concentración mínima de la sustancia activa.
Concentración máxima de cada impureza mayor o igual a CERO COMA UNO POR CIENTO PESO EN PESO (0,1 %p/p).
Concentración máxima de impurezas relevantes: el límite establecido por la normativa vigente y de no existir, el límite de detección del método utilizado.
La fracción no identificada de la sustancia activa no podrá superar el DOS POR CIENTO (2 %) por lote y deberá componerse solo de impurezas con concentración inferior al CERO COMA UNO POR CIENTO PESO EN PESO (0,1 %p/p), excluyendo impurezas relevantes.
La concentración declarada se basará en el análisis de muestras representativas de al menos CINCO (5) lotes de síntesis, asegurando que en todos los casos la concentración obtenida sea igual o superior al límite inferior establecido.
La concentración mínima de sustancia activa se determinará mediante un análisis estadístico, por ejemplo, la media menos TRES (3) desvíos estándar. Si el valor declarado es inferior, deberá justificarse el criterio estadístico o la razón para su reducción.
La concentración máxima de impurezas se obtendrá con un análisis similar, tomando la media más TRES (3) desvíos estándar. Si el valor declarado es superior, deberá explicarse el criterio utilizado o la justificación del aumento.
c - Estudio y cuantificación en CINCO (5) lotes
Debe presentarse un estudio completo de al menos CINCO (5) lotes de síntesis independientes, realizado por un laboratorio conforme a las Buenas Prácticas de Laboratorio (BPL), debidamente firmado y fechado. Si los sponsors del estudio no coinciden con la empresa registrante, deberá incluirse una autorización para el uso de la información.
El reporte debe contener:
Índice de contenidos.
Nombre y localización del establecimiento elaborador.
Cuadro resumen de resultados.
Métodos de análisis con todas las ecuaciones necesarias para reproducir los cálculos.
Resultados, incluyendo cromatogramas representativos de cada lote, del blanco y de los patrones analíticos.
En caso de usar curvas de calibración, los gráficos con títulos, ejes (con magnitudes y unidades) y ecuación de la recta.
Datos en formato de cuadro para reproducir cálculos, dentro del informe o como anexo. Cromatogramas con encabezado (fecha, hora, descripción) y reporte (áreas y tiempos de retención). Certificados de análisis de los patrones utilizados.
Certificados de análisis de los lotes analizados, emitidos y firmados por el fabricante, incluyendo nombre y dirección del establecimiento elaborador, fecha de elaboración y vencimiento. Si la dirección no figura en el certificado, deberá presentarse una declaración jurada con dicha información.
Certificado BPL del Laboratorio.
Además, el análisis de impurezas debe realizarse en los mismos lotes de síntesis, incorporando:
Un screening con detector universal para visualizar impurezas en concentraciones menores a CERO COMA UNO POR CIENTO PESO EN PESO (0,1 % p/p), ajustado a la impureza significativa de menor concentración.
Certificados de estándares con reporte de cuantificación e identificación. En caso de patrones secundarios, cuantificación contra el patrón primario y su certificado de análisis. Si se utilizan patrones de sustancias análogas, deberá presentarse una justificación técnica demostrando la analogía molecular.
d - Análisis de identidad de la sustancia activa e impurezas
La identidad de la sustancia activa deberá confirmarse mediante determinaciones analíticas que permitan establecer fehacientemente su estructura química y, si corresponde, su configuración molecular. Para uno de los CINCO (5) lotes analizados, se presentarán al menos DOS (2) espectros entre: Espectroscopía Infrarroja (IR), Resonancia Magnética Nuclear (RMN) y Espectrometría de Masas (MS). Se incluirá una discusión clara y concisa sobre la interpretación de los espectros, demostrando la identidad de la sustancia activa en grado técnico o comparándolos con los espectros del estándar analítico.
Asimismo, la identidad de todas las impurezas, o de grupos de impurezas relacionadas, deberá ser demostrada mediante análisis espectrométricos, espectroscópicos y/o químicos que permitan identificarlas inequívocamente. Cada impureza deberá estar identificada en al menos UNO (1) de los CINCO (5) lotes analizados, presentando un espectro de masa o, en caso de contar con patrones, una comparación de espectros espectroscopia ultravioleta-visible (UV-vis) y tiempos de retención. Se acompañará la información con explicaciones claras y concisas que justifiquen la identificación de cada impureza o grupo de impurezas relacionadas.
e - Métodos de análisis
El registrante debe proveer los métodos analíticos utilizados para la determinación, tanto de la sustancia activa como de las impurezas. Deberán incluir la descripción completa y los parámetros de validación respectivos (especificidad, linealidad, recuperación, precisión, límites de detección y de cuantificación en el caso de las impurezas). Además de lo especificado en los contenidos mínimos establecidos en el ítem 3, el reporte debe incluir cromatogramas representativos correspondientes a cada uno de los parámetros estudiados.
f- Proceso de síntesis
Debe proveerse la siguiente información emitida y firmada por el fabricante o la empresa registrante:
Nombre y localización del establecimiento elaborador que interviene en el proceso.
Caracterización general del proceso: deberá consistir en una descripción completa y detallada de cada una de las etapas que constituyen el proceso, incluyendo en forma explícita las reacciones químicas que tienen lugar en cada una de ellas y sus condiciones de presión y temperatura. Cuando corresponda, incluir otros parámetros controlados [por ejemplo, potencial de hidrógeno (pH), humedad, etcétera]. Detallar los criterios usados para dar por finalizada cada etapa de reacción.
Diagrama de flujo.
Identificación de todos los reactivos usados en el proceso, con las especificaciones requeridas.
Descripción de los equipos usados y sus características.
g.- Justificación de la presencia de impurezas
El fabricante deberá suministrar la explicación pertinente sobre el origen y la formación de las impurezas que pueden estar presentes en el producto final, especificando la estructura molecular y el nombre químico para cada una de ellas. Debe indicar las reacciones químicas que describen su formación, las que deben estar sustentadas en mecanismos reconocidos y probados, y compatibles con las reacciones que constituyen el proceso de síntesis.
Si el SENASA considera que una impureza declarada, o que sea probable que esté presente en el grado técnico puede ser considerada relevante, deberá solicitar a la empresa registrante que remita la discusión que fundamente su condición así como la concentración en que puede encontrarse.
En caso de que el sistema de vigilancia post registro de activos demuestre inconsistencias entre la información declarada y los análisis efectuados por la Dirección de Agroquímicos y Biológicos (DAyB) en las muestras oficiales, el SENASA se reserva el derecho de solicitar documentación adicional o dar de baja el registro.
h Sobre los efectos tóxicos en especies mamíferas (solo para sustancias activas equivalentes)
Si la evaluación de equivalencia química determina la presencia de nuevas impurezas relevantes o impurezas excedidas respecto a la referencia, el registrante deberá justificar dichas impurezas conforme a los procedimientos establecidos y presentar un test de mutagénesis junto con un informe toxicológico. Este deberá demostrar que los efectos subcrónicos y/o crónicos no son significativamente superiores a los de la sustancia parental, pudiendo basarse en estudios de relaciones cuantitativas estructura-actividad (QSAR) en DOS (2) parámetros toxicológicos crónicos relevantes.
Para evaluar los posibles efectos dañinos que deben consignarse en la etiqueta elemental del grado técnico y en la Hoja de Datos de Seguridad, se tomará como referencia la información de la sustancia parental una vez establecida la equivalencia, en concordancia con la versión adoptada por la Autoridad Competente del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA) de las Naciones Unidas.
IV - CUERPO TÉCNICO
SOBRE LAS PROPIEDADES FÍSICAS Y QUÍMICAS:
Aspecto
Estado físico
Color
Densidad
Presión de vapor
Solubilidad en agua
Solubilidad en disolventes orgánicos
Punto de inflamación
Propiedades oxidantes
Potencial de hidrógeno (pH), si la sustancia activa grado técnico es soluble o dispersable en agua.
Para el caso de sustancias activas grado técnico equivalentes, el cuerpo técnico debe incluir también la siguiente información:
SOBRE LA IDENTIDAD:
Fabricante.
Nombre común: aceptado por la Organización Internacional de Estandarización (ISO), o equivalente (si está disponible).
Nombre químico: aceptado o propuesto por IUPAC o CAS (si está disponible).
Número CAS, CIPAC y/o número de código experimental.
Fórmula empírica, peso molecular.
Fórmula estructural.
Grupo químico.
Concentración de sustancia activa.
SOBRE LOS ASPECTOS RELACIONADOS A SU USO.
1. Organismos nocivos controlados.
2. Modo de acción sobre los organismos nocivos y sobre las plantas.
3. Ámbito de aplicación previsto.
4. Condiciones fitosanitarias y ambientales para ser usado.
SOBRE LOS MÉTODOS ANALÍTICOS (para la determinación de residuos en cultivos tratados, subproductos de origen vegetal, alimentos procesados, suelo y agua)
Se incluirá la tasa de recuperación y los límites de sensibilidad metodológica.
Estos métodos serán requeridos si los usos propuestos para el producto requieren el establecimiento de una tolerancia.
Métodos analíticos para la determinación en el aire: Estos serán requeridos para productos volátiles. Métodos analíticos para la determinación en tejidos y fluidos animales o humanos: Estos métodos serán requeridos cuando las plantas tratadas se destinen a la alimentación animal.
V- RESIDUOS EN PRODUCTOS TRATADOS (ÚNICAMENTE REQUERIDO PARA SUSTANCIAS ACTIVAS DE GRADO TÉCNICO NUEVAS y AMPLIACIONES DE NUEVO USO)
Los estudios serán requeridos si los usos propuestos exigen el establecimiento de una tolerancia. El cálculo del Límite Máximo de Residuos (LMR) se realizará utilizando la calculadora de la OCDE, basada en los datos disponibles.
a. Metabolismo en Vegetales (para sustancias activas nuevas)
Debe identificarse la degradación y reacción de metabolitos en plantas o productos tratados. Se requiere un estudio de metabolismo en un cultivo representativo de cada categoría o grupo de cultivos para los que se propone el uso, conforme a las Directrices de la OCDE u otras reconocidas. Para extrapolar el metabolismo a todos los grupos, se realizarán estudios en al menos TRES (3) de las CINCO (5) categorías de cultivos; si los resultados son comparables, no serán necesarios estudios adicionales.
b. Comportamiento de Residuos (para sustancias activas nuevas y ampliaciones de uso en nuevos cultivos, incremento de dosis, reducción del tiempo de carencia)
Para usos que requieran la fijación de tolerancias, se realizarán ensayos de residuos en cultivos para establecer un LMR. Estos estudios deberán cumplir con Buenas Prácticas de Laboratorio (BPL) en sus fases analíticas y de campo. En caso de requerirse estudios locales, la fase de campo deberá realizarse en la REPÚBLICA ARGENTINA con certificación BPL.
Para aquellos usos que requieran la fijación de tolerancias, se requerirán ensayos de residuos en cultivos a fin de establecer un Límite Máximo de Residuos para el cultivo o grupo de cultivos para el que se requiera autorizar el uso de la sustancia activa.
Los estudios de residuos de productos fitosanitarios deben ser realizados por entidades que hayan certificado el cumplimiento de Buenas Prácticas de Laboratorio (BPL), tanto en sus fases analíticas, como de campo.
Para aquellos casos en los que se requiera un número mínimo de estudios de residuos locales, la fase de campo deberá realizarse en el territorio Argentino por entidades que hayan certificado el cumplimiento de BPL.
El plan de ensayos deberá ajustarse a este Manual y los ensayos de campo en Argentina deberán emplear variedades representativas en la región evaluada. Para ensayos realizados en la misma campaña, los lotes experimentales deben distanciarse al menos VEINTE (20) kilómetros y tener un desfase de QUINCE (15) días.
Los ensayos deben respetar la práctica agrícola crítica indicada en la etiqueta del producto formulado: dosis máxima, número de aplicaciones, intervalo entre aplicaciones y período de carencia. Previa justificación técnica, se aceptará una variación en más o en menos de un VEINTICINCO POR CIENTO (±25 %) en un parámetro de la práctica agrícola crítica y el criterio de proporcionalidad de la dosis máxima, siguiendo directrices de la OCDE.
Cuando se requieran curvas de degradación, estas deberán incluir al menos TRES (3) puntos y abarcar el período de carencia. Si el residuo es menor al Límite de Cuantificación (LOQ), no será necesario realizar curvas de degradación.
Los ensayos destinados a establecer un periodo de seguridad (usos en postcosecha), no requerirán curvas de disipación, pero las muestras deben tomarse dentro del período de seguridad propuesto. El producto formulado utilizado en el ensayo debe caracterizarse bajo principios BPL, con Certificado de Análisis incluido en el informe. Las muestras deben analizarse en un máximo de TREINTA (30) días tras la recolección; en caso contrario, deberá presentarse un estudio de estabilidad de la molécula y sus metabolitos en una matriz representativa.
Productos exentos de fijación de LMR:
• Tratamiento de semillas y órganos de propagación no destinados al consumo.
• Aplicaciones en cultivos florales, ornamentales o áreas no cultivadas.
• Arbusticidas de acción tópica, preservadores de madera y hormiguicidas no aplicables al vegetal.
• Feromonas, atractivos, repelentes, rodenticidas y coadyuvantes.
• Biocontroladores
• Otros productos definidos con base técnica y científica.
c. Residuos en Cultivos Rotacionales (Nuevas sustancias activas - Condicionalmente Requeridos)
Si la sustancia activa se usa en cultivos anuales y sus metabolitos persisten en el suelo, pueden exigirse estudios de metabolismo en cultivos de rotación.
Si estos estudios indican residuos de la sustancia activa o metabolitos MAYOR A CERO COMA CERO UN MILIGRAMOS POR KILOGRAMO (> 0,01 mg/kg) en alimentos o MAYOR O IGUAL A CERO COMA CERO CINCO MILIGRAMOS POR KILOGRAMO (> 0,05 mg/kg) en forrajes, se seguirán directrices de la OCDE. Se aceptarán estudios realizados en otros países.
VI - EFECTOS TÓXICOS EN ESPECIES MAMÍFERAS. (ÚNICAMENTE REQUERIDO PARA SUSTANCIAS ACTIVAS DE GRADO TÉCNICO NUEVAS)
Se deberá aplicar un enfoque basado en toda la información disponible (peso de la evidencia) para determinar si un estudio estándar, un estudio con parámetros adicionales o un método alternativo al uso de animales, permite evaluar adecuadamente el peligro para la salud humana o justificar una exención de estudios específicos.
Los estudios deberán seguir las Test Guidelines y Guidance Documents de la OCDE u otros reconocidos internacionalmente.
a - TOXICIDAD AGUDA
Se requerirá el estudio de toxicidad oral aguda (DL50 oral) en todos los casos, excepto cuando la sustancia activa de grado técnico sea un gas o altamente volátil.
El estudio de toxicidad cutánea aguda (DL50 cutánea) será obligatorio al momento de la inscripción definitiva, salvo que el producto formulado sea un gas, altamente volátil o corrosivo para la piel, o presente un pH inferior a DOS (2) o superior a ONCE COMA CINCO (11,5).
El estudio de toxicidad aguda por inhalación (LC50 inhalación) será requerido cuando la sustancia activa de grado técnico tenga una presión de vapor superior a 10-2 Pa a 20 °C, sea un polvo con una proporción significativa de partículas con diámetro menor a 50 p,m [más del UNO POR CIENTO (1 %) en peso] o se incluya en productos en polvo o que se apliquen por pulverización.
El estudio de irritación cutánea in vivo no deberá realizarse cuando la sustancia activa de grado técnico sea un gas o altamente volátil, sea corrosiva para la piel, presente un pH inferior a DOS (2) o superior a ONCE COMA CINCO (11,5), cuente con información suficiente que permita clasificarla como corrosiva para la piel o irritante ocular, o haya sido clasificada como muy tóxica por vía dérmica.
El estudio de irritación ocular será obligatorio, salvo que la sustancia activa de grado técnico sea corrosiva o severamente irritante para la piel, presente un pH inferior a DOS (2) o superior a ONCE COMA CINCO (11,5) o haya sido clasificada como muy tóxica por vía dérmica.
El estudio de sensibilización cutánea será requerido, excepto cuando la sustancia activa sea un sensibilizante conocido.
En cuanto a la mutagenicidad, será obligatorio realizar un estudio in vitro en el momento del registro experimental. Si la empresa registrante no dispone de estudios toxicológicos in vivo, podrá presentar estudios alternativos in vitro, in silico u otros métodos reconocidos por autoridades regulatorias internacionales con convergencia normativa entre la REPÚBLICA ARGENTINA, para el registro de productos fitosanitarios. En estos casos, deberá solicitarse su aceptación mediante una nota con una justificación técnico-científica del uso de dichos estudios.
b - TOXICIDAD SUBCRÓNICA (corto plazo/medio plazo)
- Cuando esté disponible, se deberán comunicar los estudios de toxicidad oral acumulativa con una duración de VEINTIOCHO (28) días.
- El estudio de toxicidad oral a corto plazo en roedores deberá realizarse con una duración de NOVENTA (90) días, utilizando ratas como especie de prueba, salvo que se justifique el uso de otra especie. Para no roedores, se deberá presentar un estudio de toxicidad de NOVENTA (90) días en perros. En ambos casos, deberán evaluarse el potencial neurotóxico, los efectos inmunotóxicos, la genotoxicidad a través de la formación de micronúcleos y los posibles efectos sobre el sistema hormonal.
- El estudio de toxicidad inhalatoria con exposición repetida durante VEINTIOCHO (28) o NOVENTA (90) días será requerido condicionalmente para sustancias activas volátiles con una presión de vapor superior a 10“2 Pascales, cuando sea necesario para refinar la evaluación del riesgo ocupacional.
El estudio de toxicidad dérmica con administración repetida durante VEINTIÚN (21), VEINTIOCHO (28) o NOVENTA (90) días será requerido condicionalmente para el refinamiento de la evaluación del riesgo ocupacional, en caso de que los estudios de toxicología aguda dérmica o los valores por defecto de penetración dérmica no sean suficientes para su determinación.
c - TOXICIDAD CRÓNICA A LARGO PLAZO Y CARCINOGENICIDAD.
Se requerirá un estudio de toxicidad oral a largo plazo y un estudio de carcinogenicidad a largo plazo de la sustancia activa, utilizando ratas como especie de prueba; cuando sea posible, estos estudios se combinarán.
Las duraciones mínimas de estudio aceptables son:
Estudio de carcinogenicidad en ratones: 18 meses.
Estudio combinado toxicidad oral a largo plazo y carcinogenicidad en ratas: 12 - 24 meses.
Se requerirá un segundo estudio de carcinogenicidad de la sustancia activa utilizando ratón como especie de ensayo, a menos que pueda justificarse científicamente que no es necesario. En tales casos, se pueden utilizar modelos alternativos de carcinogenicidad validados científicamente.
Se deben remitir los datos experimentales, incluyendo la determinación del posible mecanismo de carcinogenicidad implicado y su relevancia para los seres humanos, cuando se considere que el modo de acción de la carcinogenicidad no es genotóxico.
d.- MUTAGENICIDAD
Los estudios de mutagenicidad in vitro deberán realizarse según corresponda, incluyendo al menos uno de los siguientes ensayos: ensayo bacteriano para mutación genética, prueba combinada para aberraciones cromosómicas estructurales y numéricas en células de mamífero, o prueba de mutación genética en células de mamíferos.
Los estudios de mutagenicidad in vivo en células somáticas solo serán requeridos si al menos uno de los estudios in vitro arroja un resultado positivo. En tal caso, deberá realizarse al menos un estudio in vivo que evalúe específicamente el efecto de genotoxicidad identificado en los ensayos in vitro. Si se requiere un estudio de micronúcleos in vivo, este podrá integrarse dentro de un estudio de dosis repetida.
La necesidad de estudios in vivo en células germinales se evaluará caso por caso, en función de los resultados obtenidos en células somáticas, considerando la toxicocinética de la sustancia, su uso y la exposición anticipada.
e - EFECTOS SOBRE LA REPRODUCCIÓN
Para evaluar los efectos sobre la reproducción, se recomienda la realización de un estudio combinado de toxicidad reproductiva en ratas, utilizando un estudio de dos generaciones o de una generación extendida como protocolo base. Este estudio deberá incluir criterios adicionales de valoración y evaluaciones funcionales en animales inmaduros.
Los estudios de toxicidad del desarrollo serán obligatorios y deberán realizarse en ratas y conejos por vía oral.
f - METABOLISMO EN MAMÍFEROS
Se deberán realizar estudios in vitro comparativos del metabolismo de la sustancia activa entre especies animales y humanos.
Los estudios in vivo en mamíferos deberán proporcionar información sobre la cinética de la sustancia activa y sus metabolitos en especies relevantes, considerando:
Una dosis oral única en niveles de dosis baja y alta.
Una dosis intravenosa o, si está disponible, una dosis oral única con evaluación de la excreción biliar en nivel de dosis baja.
Una dosis repetida.
Para el refinamiento de la evaluación de riesgo ocupacional, podrá requerirse un estudio de penetración dérmica in vitro en piel humana a fin de determinar la biodisponibilidad por vía dérmica.
Asimismo, deberá incluirse una descripción detallada de las rutas metabólicas de la sustancia activa y sus metabolitos.
g - ESTUDIOS DE NEUROTOXICIDAD EN ROEDORES
Los estudios serán condicionalmente requeridos a partir de un enfoque basado en el peso de la evidencia, considerando que:
La sustancia causa efectos neurológicos en animales adultos (es decir, signos clínicos de neurotoxicidad, neuropatología, alteraciones funcionales o de comportamiento).
La sustancia causa efectos neurológicos en animales en desarrollo, después de la exposición pre y posnatal (es decir, malformaciones del sistema nervioso o neuropatía, cambios de peso cerebral en la descendencia, alteraciones funcionales o de comportamiento en la descendencia). la sustancia evoque un mecanismo que está asociado con efectos adversos sobre el desarrollo del sistema nervioso, en comparación con neurotóxicos conocidos, respuestas de neurorreceptores o neurotransmisores alterados.
Se recomienda el uso de un enfoque combinado que utilice el estudio de reproducción de DOS (2) generaciones en roedores o de UNA (1) generación extendida, como protocolo básico para la evaluación de efectos de neurotoxicidad y evaluaciones funcionales en animales inmaduros.
h- OTROS ESTUDIOS TOXICOLÓGICOS
Cuando los metabolitos de la sustancia activa generados en plantas, productos animales, suelo o agua subterránea difieran de aquellos identificados en los estudios toxicológicos en animales, o se detecten en bajas proporciones en estos últimos, se requerirán estudios adicionales. La necesidad de estos estudios se evaluará caso por caso, considerando la cantidad y la estructura química del metabolito en relación con la sustancia parental.
En lo que respecta a la Información Médica Obligatoria, se deberá presentar información sobre: Diagnóstico y síntomas de intoxicación.
Observaciones sobre sensibilización y alergización.
Efectos tóxicos de los metabolitos provenientes de vegetales tratados.
Información médica complementaria, cuando esté disponible.
Elaboración de una ficha médica definitiva.
i - INFORMACIÓN CON RESPECTO A LA SEGURIDAD
Se deberán establecer procedimientos específicos para la destrucción de la sustancia activa y su descontaminación, así como la evaluación de posibles métodos de recuperación, neutralización e incineración controlada, indicando las condiciones en que debe realizarse.
Asimismo, deberá detallarse la depuración de aguas y los métodos recomendados para la manipulación, almacenamiento, transporte y respuesta ante incendios o derrames, incluyendo información sobre los productos de reacción y gases de combustión generados en caso de incendio. Finalmente, se deberá proporcionar información sobre los equipos de protección individual adecuados para minimizar los riesgos asociados a la exposición a la sustancia activa.
j.- EVALUACIÓN TOXICOLÓGICA
Para la identificación de peligros, deberá determinarse el Nivel de Efecto Adverso No Observable (NOAEL) con base en los estudios más representativos de los escenarios de uso del producto en Argentina, tanto para la evaluación de riesgos ocupacionales como dietarios. Se considerará el estudio toxicológico crónico más sensible y representativo de la vía de exposición evaluada.
En la evaluación de riesgo dietario, deberá establecerse la Ingesta Diaria Admisible (IDA) con base en el NOAEL del estudio toxicológico de largo plazo más sensible y representativo por vía oral, aplicando un factor de incertidumbre técnicamente justificado. Además, salvo prueba científica en contrario, deberá definirse una Dosis de Referencia Aguda (ARfD) basada en estudios de corto plazo por vía oral.
Asimismo, deberá realizarse la clasificación de peligros a la salud y de los peligros físicos de la sustancia activa conforme a la versión adoptada por la Autoridad Competente del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA) de las Naciones Unidas.
Definición del Residuo en Material Tratado:
Deberá proporcionarse la definición de residuos con fines de análisis de riesgo y monitoreo.
La inclusión de metabolitos o productos de degradación en la definición para evaluación del riesgo dependerá de su perfil toxicológico y su magnitud, cuando superen el DIEZ POR CIENTO (10 %) del total de residuos radioactivos.
Para la definición de residuos con fines de monitoreo, debe ser inequívoca y simple, preferentemente basada en un único compuesto (generalmente el compuesto parental), para todas las commodities.
Evaluación de la Exposición y Caracterización del Riesgo Dietario:
La evaluación del riesgo dietario, tanto crónico como agudo, se realizará conforme a metodologías internacionalmente aceptadas, considerando los ensayos de residuos supervisados para la sustancia activa, los valores de referencia toxicológicos (IDA y ARfD) y las estadísticas de consumo de la población argentina o una dieta representativa de la región.
Se considerará aceptable la exposición alimentaria crónica cuando la Ingesta Diaria Estimada Nacional sea inferior al CIEN POR CIENTO (100 %) de la IDA. De igual manera, la exposición alimentaria aguda será aceptable si la Ingesta Nacional Estimada de corto plazo es menor al CIEN POR CIENTO (100 %) de la ARfD.
Evaluación de la Exposición y Caracterización del Riesgo Laboral:
La evaluación de la exposición podrá basarse en estudios específicos o estimaciones mediante datos genéricos y modelos de simulación aceptados internacionalmente. Los escenarios de exposición deberán considerar las prácticas laborales, el uso y disponibilidad del equipo de protección personal, en concordancia con la práctica agrícola recomendada en la etiqueta.
El criterio de aceptabilidad del riesgo estará determinado por el Margen de Exposición (MOE), calculado como el cociente entre el NOAEL y la exposición estimada. Un MOE superior a CIEN (100) se considerará como riesgo aceptable.
En aquellos casos en que el valor de CIEN (100) pueda variar asociado a razones como relevancia de un estudio de ruta específica, ausencia de NOAEL u otras causas atendibles y válidas, deberá justificarse y validarse científicamente si se utiliza otro valor como límite de preocupación diferente a CIEN (100).
Período de Reingreso al Lote Tratado:
Cuando corresponda, se deberá realizar una evaluación de riesgo para el trabajador de reingreso al lote tratado. Para su determinación, deberán presentarse datos sobre toxicidad aguda dérmica, potencial irritante dérmico y ocular, y sensibilización dérmica del principio activo o del producto formulado.
VII - EFECTOS TÓXICOS SOBRE OTRAS ESPECIES (ÚNICAMENTE REQUERIDO PARA SUSTANCIAS ACTIVAS DE GRADO TÉCNICO NUEVAS).
Los estudios deberán realizarse conforme a las Test Guidelines y Guidance Documents de la OCDE u otras referencias internacionalmente reconocidas.
Para evaluar los efectos en aves, se deberán realizar estudios de toxicidad oral aguda, toxicidad dietaria subcrónica y efectos en la reproducción, utilizando especies validadas como faisán, codorniz o pato silvestre.
En cuanto a peces, se requerirá la evaluación de toxicidad aguda y crónica en especies como trucha arco iris o carpa, así como estudios de bioacumulación, los cuales serán exigidos de manera condicional para sustancias con un Log Kow superior a TRES (3).
Para invertebrados acuáticos, se deberá evaluar la toxicidad aguda y crónica en Daphnia magna. Asimismo, se evaluarán los efectos en organismos acuáticos mediante estudios sobre el crecimiento de algas, utilizando Selenastrum capricornutum u otra especie validada, y la inhibición del crecimiento de plantas acuáticas, tomando como referencia Lemna sp.
El impacto sobre invertebrados terrestres deberá analizarse mediante estudios de toxicidad aguda y crónica en abejas. Para las abejas adultas, se evaluará la toxicidad oral y por contacto (DL50); mientras que la toxicidad oral aguda en larvas y los estudios crónicos en adultos y larvas se determinará será requerido condicionalmente cuando la sustancia activa sea sistémica en el vegetal y presente un DL50 inferior a ONCE (11) miligramos/abeja. Quedan exceptuados los formulados de uso exclusivo en poscosecha, invernáculos o cebos.
Además, se deberá evaluar la toxicidad aguda en artrópodos benéficos en especies validadas como Aphidius rhopalosiphi, Chrysoperla carnea u Orius laevigatus. Para lombrices de tierra, se evaluará la toxicidad aguda y el impacto en la reproducción en Eisetia foetida u otra especie validada.
Asimismo, se analizarán los efectos sobre colémbolos en el suelo, particularmente en Folsomia candida y Folsomia fimetaria, mediante estudios de reproducción.
Finalmente, se evaluará la toxicidad en microorganismos del suelo, enfocándose en aquellos involucrados en procesos de nitrificación.
VIII - EFECTOS SOBRE EL MEDIO ABIÓTICO (ÚNICAMENTE REQUERIDO PARA SUSTANCIAS ACTIVAS DE GRADO TÉCNICO NUEVAS).
Comportamiento en el suelo.
Se requerirán estudios en al menos TRES (3) tipos de suelos patrón. Las evaluaciones deben conducirse considerando concentraciones de la sustancia equivalentes a la práctica agrícola crítica que se quiere registrar. Los suelos patrón deben ser representativos de los suelos agrícolas, pueden incluir uno de textura gruesa, otro de textura franca y otro de textura fina.
Se debe determinar la tasa de degradación, hasta el NOVENTA POR CIENTO (90%), y vías de degradación incluida la identificación de: Procesos que intervienen; Metabolitos y productos de degradación (Degradación Aeróbica y Anaeróbica/ Fotólisis); Absorción y desorción y movilidad de la sustancia activa y si es relevante, de sus metabolitos; Magnitud y naturaleza de los residuos remanentes.
Comportamiento en el agua y en el aire.
Se debe determinar la tasa de degradación, hasta el NOVENTA POR CIENTO (90%), y vías de degradación. Biodegradación.
m - EVALUACIÓN DE RIESGO AMBIENTAL
Formulación del problema:
En función del patrón de uso y las formulaciones representativas a registrar, se deben determinar grupos potenciales de especies no blanco que podrían estar expuestas y requerir evaluación de riesgo.
Evaluación de la exposición ambiental
El destino ambiental de una sustancia química dependerá del patrón de uso definido en la formulación del problema, por ende se deben considerar tanto los factores, como el método de aplicación, los cultivos objetivo, la época del año en que se propone la aplicación de la sustancia activa y el área geográfica en la que se utilizará.
Como producto final se deberá definir la concentración ambiental estimada (CAE), definida como la cantidad de sustancia activa y sus metabolitos toxicológicamente relevantes en agua, suelo o sedimento y aire, como resultado del uso propuesto. En ausencia de modelos de exposición locales, se podrán utilizar los procedimientos desarrollados por la Agencia de Protección Ambiental de Estados Unidos (US-EPA).
La evaluación de riesgos ambientales será desarrollada como un proceso escalonado, de diferentes fases de refinamiento de la información, tendiente a reducir la incertidumbre en la toma de decisiones.
Para nuevas sustancias activas, las fases serán:
Fase 1: Estudios de laboratorio estándar requeridos en esta norma: propiedades fisicoquímicas, destino ambiental, toxicidad aguda y/o crónica. En esta fase de la evaluación de riesgos, se utiliza el llamado enfoque de 'peor caso', el cual combina los valores más altos de exposición (máxima biodisponibilidad), con los niveles más altos de toxicidad (especie sensible).
Fase 2: Modelos de estimación de la distribución y degradación ambiental del compuesto parental y sus metabolitos toxicológicamente relevantes en suelo, agua y aire. La elección estará determinada por las propiedades individuales y los usos de una sustancia. En ausencia de modelos de exposición locales, se deberán utilizar los procedimientos de estimación ambiental predicha desarrollados por la Agencia de Protección Ambiental de Estados Unidos (US-EPA).
Fase 3: Estudios de semi-campo o de campo simulados, en caso de que el riesgo de un producto no pueda evaluarse suficientemente luego de las fases de evaluación 1 y 2, particularmente para sustancias que son relativamente persistentes o exhiben una alta movilidad. Se deberá acordar con la Autoridad Competente el protocolo a utilizar.
Caracterización del riesgo
Como procedimiento para integrar los resultados del análisis de la exposición con los efectos ecotoxicológicos adversos potenciales, puede utilizarse el método determinístico que utilice la aproximación de los COCIENTES DE RIESGO (RQ), calculados como el cociente entre los CAE y los valores de ecotoxicidad aguda o crónica, según corresponda. Sin embargo, pueden aplicarse otras metodologías aceptadas internacionalmente, a fin de establecer perfil ecotoxicológico de la sustancia en estudio.
DECLARACION DE NUEVOS ESTABLECIMIENTOS ELABORADORES
La solicitud de registro de una sustancia activa grado técnico registrada previamente por el solicitante, proveniente de un nuevo establecimiento elaborador debe realizarse mediante la presentación de la información y documentación requerida en el presente Anexo, Título “III. INFORMACIÓN CONFIDENCIAL (para sustancias activas nuevas y equivalentes)”.
La Autoridad Competente puede dar curso a la gestión de la solicitud, aprobación y registro una vez admitida la presentación de la información y documentación requerida.
B - PRODUCTOS FORMULADOS:
Además de los productos formulados de síntesis este registro también contempla los siguientes supuestos:
Los formulados que entre sus componentes contengan productos sintéticos solos o en mezcla con sustancias de origen biológico que por su forma de uso generen una respuesta en la conducta o en la fisiología del organismo plaga que se desee controlar, de aquí en adelante serán llamados semioquímicos.
Los formulados que contengan bactericidas, fungicidas y/o insecticidas destinados a prevenir, detener y/o eliminar el ataque de bacterias, hongos y/o insectos que afecten a las maderas en sus distintas formas y serán denominados de aquí en adelante preservadores para la madera.
Los formulados que sean producidos en base a agentes microbianos de ocurrencia natural, o introducidos en el ambiente, para la prevención y/o el control de organismos considerados plaga de la agricultura; sustancias bioquímicas (extractos de origen vegetal, animal o microbiológico). Invertebrados para el control biológico de plagas de la agricultura.
En los casos que se desee inscribir un producto formulado, se deberá presentar la documentación descrita a continuación. dada la diversidad de características asociadas al origen, naturaleza y aptitud de uso, los y preservadores de madera serán tratados en el apartado de LOS REQUISITOS Y PROCEDIMIENTOS DE PRODUCTOS DE TRATO DIFERENCIADO (PTD).
I - INFORMACIÓN ADMINISTRATIVA
Formulario de solicitud de registro dependiendo el tipo de formulado firmado por el Apoderado o Representante Legal y el Responsable Técnico con incumbencia profesional en la materia.
Proyecto de marbete según normativa vigente.
Hoja de Datos de Seguridad, de acuerdo a la versión del Manual de las Naciones Unidas del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA), adoptada en la presente norma.
Patrones: en caso de que el SENASA así lo requiera, a los fines de control y vigilancia, la empresa registrante deberá aportar los patrones y metodología analítica de la/s sustancia/s activa/s, en las condiciones que establezca la Dirección General de Laboratorios y Control Técnico (DGLCyT) del SENASA.
Sistema de gestión de envases vacíos aplicado en cumplimiento de la Ley vigente.
Formulario II (condicionalmente requerido).
Autorización a terceros a hacer uso del registro de un principio activo grado técnico para el registro de un producto formulado.
II - INFORMACIÓN CONFIDENCIAL
a.- Certificado de Origen del producto formulado.
El Certificado de Origen debe ser original y emitido por el establecimiento elaborador.
Para establecimientos no ubicados en la REPÚBLICA ARGENTINA, para cada uno de los orígenes a registrar.
Debe incluir:
Identificación de la/s sustancia/s activa/s; Concentración declarada para cada sustancia activa Tipo de formulación armonizada; Nombre y domicilio real del establecimiento formulador.
b. - Declaración de composición cuali-cuantitativa del producto formulado firmada por el establecimiento elaborador o por la empresa registrante con carácter de declaración jurada y que deberá contener:
Nombre químico según criterios internacionales (IUPAC o CAS) y número CAS (cuando está disponible) de la/s sustancia/s activa/s y demás componentes.
Contenido nominal de la(s) sustancia(s) activa(s), en base CIEN POR CIENTO (100 %) de pureza, expresado en porcentaje en masa (% p/p) para los formulados sólidos o gaseosos, y en porcentaje masa/volumen (% p/v) para los formulados líquidos.
Contenido nominal, naturaleza química y función específica de cada uno de los componentes restantes incluidos en la formulación.
Tipo de formulación armonizada de acuerdo con la Norma IRAM 12074.
Se presentará una sola declaración de composición cuali-cuantitativa representativa para todas las plantas para las que se solicita el registro en la que debe figurar la razón social y dirección real de cada uno de los establecimientos fabricantes.
c. - Estudio de UN (1) lote
Se debe presentar el análisis de una muestra representativa de UN (1) lote de formulación para la cuantificación de la(s) sustancia(s) activa(s), proveniente de una de las plantas para la que se solicita el registro o del Laboratorio de desarrollo. Los valores de concentración informados para la(s) sustancia(s) activa(s) deberán cumplir con los límites establecidos en la Norma IRAM 12054, en función del contenido nominal declarado.
En caso de que el sponsor del estudio no coincida con la empresa registrante se deberá remitir una autorización del sponsor para la utilización de la información.
Deberá presentarse el estudio completo, de acuerdo con los principios de Buenas Prácticas de Laboratorio, convenientemente firmado y fechado.
Se establecen los siguientes contenidos mínimos para el reporte del estudio de 1 lote:
Índice de contenidos
Nombre y domicilio real del establecimiento elaborador Cuadro de resumen de resultados
Métodos de análisis, que incluya todas las ecuaciones necesarias para reproducir los cálculos realizados
Resultados
En caso de usar curvas de calibración incluir los gráficos identificando título y ejes (con magnitud y unidades). Además debe incluir la ecuación de la recta. Cuando corresponda incluir los cálculos de densidad. Se deben incluir en formato de cuadro los datos necesarios para poder reproducir los cálculos realizados.
Los cromatogramas representativos del lote y del patrón analítico, con encabezado (con fecha, hora, descripción) y reporte (áreas y tiempo de retención). Certificados de análisis de todos los patrones utilizados. Cuando sean generados por el fabricante o el laboratorio deben incluir el reporte de cuantificación e identificación. En caso de patrones secundarios incluir la cuantificación contra el patrón primario junto a su certificado de análisis. Certificado de análisis del lote analizado emitido y firmado por el formulador que incluya nombre y dirección del establecimiento formulador y fecha de elaboración y vencimiento del lote. De no disponer en el certificado de análisis la dirección del establecimiento elaborador, se debe presentar una declaración jurada indicando dicha información acompañando el certificado. Constancia BPL OECD del Laboratorio
Acreditación BPL del estudio.
b.- Proceso de formulación
Para cada proceso de formulación debe proveerse la siguiente información emitida y firmada por el fabricante o la empresa registrante:
Nombre y domicilio real de los establecimientos formuladores que interviene en el proceso. Caracterización general del proceso: deberá consistir en una descripción completa y detallada de cada una de las etapas que constituyen el proceso, especificando qué componentes son agregados en cada una y qué operación se realiza.
Identificación de los ingredientes usados para formular el producto.
Descripción de los equipos usados, con sus especificaciones.
Descripción de las condiciones que se controlan en cada etapa del proceso y los parámetros de control de calidad del producto terminado.
III - CUERPO TÉCNICO
Este cuerpo técnico se presentará con la documentación dispuesta por temas, a saber:
a- COMPOSICIÓN
Contenido de sustancia(s) activa(s), grado técnico, expresado en % p/p o % p/v según corresponda Métodos de análisis para la determinación del contenido de la(s) sustancia(s) activa(s).
Para el caso de preservadores de la madera: deberá expresarse en óxidos cualesquiera fueran las sustancias que los integran, (como lo establece la norma IRAM). Y adicionalmente mencionar el método de aplicación: pincelado, aspersión, inmersión, prolongada, momentánea, breve, inyección, baño caliente-frío.
b - PROPIEDADES DEL PRODUCTO FORMULADO
Propiedades Físicas y Químicas.
Aspecto: Tipo de Formulación Armonizada; Color
Estabilidad en el almacenamiento a baja temperatura y acelerada a alta temperatura
Densidad relativa Inflamabilidad
Acidez/Alcalinidad y potencial de hidrógeno (pH).
Propiedades físicas relacionadas con su uso.
Humectabilidad: Para polvos dispersables o mojables.
Persistencia de la espuma: Para formulados que se aplican con agua. Suspensibilidad: Para gránulos dispersables (WG), polvos mojables: (WP), suspensiones concentradas: (SC), Suspensión de encapsulado (CS) Análisis granulométrico en húmedo: Para los polvos mojables, las suspensiones concentradas (SC, FS), Suspoemulsión (SE).
Análisis granulométrico en seco: Para gránulos y polvos. Estabilidad de la emulsión: Para concentrados emulsionables. Corrosividad.
Incompatibilidad con otros productos: Con otro fitosanitario y/o fertilizante. Densidad: Para sólidos y líquidos.
Punto de inflamación: Para líquidos.
Viscosidad: Para aceites, suspensiones y concentrados emulsionables. Índice de sulfonación: (Residuo no Sulfonable) Para aceites y aceites emulsionables destinados a frutales u ornamentales).
Dispersión: Para gránulos dispersables.
Desprendimiento de gases: Para gránulos generadores de gas. Soltura o fluidez: Para polvos secos.
Índice de iodo: Índice de Iodo y de Saponificación. Sólo para aceites vegetales, no para losaceites minerales.
Velocidad de liberación. Para suspensiones acuosas encapsuladas (CS) y para la formulación mixta (ZC), mezcla de suspensiones encapsuladas y suspensiones concentradas (CS + SC).
Presión de vapor, si corresponde en función de la volatilidad.
IV. ENSAYOS DE EFICACIA AGRONÓMICA Y FITOTOXICIDAD
Aquellas recomendaciones de usos que no tengan antecedentes en Argentina o las modificaciones de los usos críticos ya autorizados, deberán respaldar la solicitud mediante informes de los ensayos de eficacia agronómica y fitotoxicidad.
V ETIQUETADO
El etiquetado deberá ajustarse a la normativa vigente, y conforme a la versión del Manual de las Naciones Unidas del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA) adoptada por la Autoridad Competente del Registro Nacional.
VI. ENVASES Y EMBALAJES PROPUESTOS.
a.- Envases.
Tipo(s)
Material(es)
Capacidad(es)
b.-Embalajes.
Tipo
Material
Procedimientos para la descontaminación y destino final de los embalajes.
VII - EFECTOS TÓXICOS EN ESPECIES MAMÍFERAS
En el caso que la empresa registrante no disponga de estudios toxicológicos in vivo, como alternativa de reemplazo, reducción o refinamiento de las pruebas con animales requeridas en este apartado, podrán presentarse estudios in vitro, siempre y cuando, dichos protocolos y ensayos hayan sido reconocidos por autoridades regulatorias internacionales para el Registro Nacional de Productos Fitosanitarios. En tales casos, el registrante deberá solicitar mediante una nota la aceptación de dichos estudios, mediante una justificación técnico-científica de su uso.
Se aplicarán los criterios de las directrices de la OCDE para justificar la no conducción de estudios in vivo.
a.- Toxicidad aguda.
Oral. Toxicidad oral aguda (DL50 oral)
Este estudio se requerirá en todos los casos excepto si el producto formulado es un gas o es altamente volátil.
Dérmica. Toxicidad cutánea aguda (DL50 cutánea)
Este estudio se requerirá a menos que:
El producto formulado es un gas o es altamente volátil. El producto formulado es corrosivo para la piel o presenta un pH menor a DOS (2) o mayor a ONCE COMA CINCO (11,5).
Inhalatoria. Toxicidad aguda por inhalación (LC50 inhalación) Este estudio se requerirá cuando el producto formulado es un gas o es altamente volátil; o consiste en, o en las condiciones de uso darán como resultado, un material respirable (por ejemplo, gas, vapor, aerosol o partículas).
b.- Irritación cutánea y ocular.
Irritación cutánea.
El estudio in vivo no debería realizarse si: El producto formulado es un gas o es altamente volátil. El producto formulado es corrosivo para la piel o presenta un pH menor a DOS (2) o mayor a ONCE COMA CINCO (11,5)
Existe información disponible que indica que satisface los criterios para ser clasificada como corrosiva para la piel o irritante ocular La sustancia es clasificada como muy tóxica por vía dermal
Irritación ocular.
Este estudio se requerirá a menos que:
El producto formulado sea corrosivo o severo irritante para la piel,
Presente un pH menor a DOS (2) o mayor a ONCE COMA CINCO (11,5)
La sustancia es clasificada como muy tóxica por vía dermal.
c.- Sensibilización cutánea.
Este estudio se requerirá a menos que el principio activo sea un sensibilizante conocido.
VIII - INFORMACIÓN MÉDICA OBLIGATORIA
Diagnóstico y síntomas de intoxicación
Tratamientos propuestos
Primeros auxilios. Antídotos
Tratamiento médico.
IX - DATOS DE LOS EFECTOS SOBRE EL AMBIENTE
a.- Toxicidad aguda en aves Estos estudios serán requeridos para todos los productos cuyos usos propuestos son en lugares abiertos. Toxicidad oral aguda (en faisán, codorniz, pato silvestre y otra especie validada).
b.- Toxicidad aguda sobre organismos acuáticos Estos estudios serán requeridos para todos los productos cuyos usos propuestos son en lugares abiertos. Concentración letal media de NOVENTA Y SEIS HORAS (96 h) en peces (en trucha arco iris, carpa y otras especies validadas).
d.- Efectos tóxicos en polinizadores e invertebrados no blanco
Este estudio será requerido si por el uso propuesto pudiere resultar una exposición de las abejas.
Toxicidad oral aguda para las abejas adultas (DL50 oral adultas).
Toxicidad por contacto agudo para las abejas adultas (DL50 contacto adulto).
X EVALUACIÓN TOXICOLÓGICA Y ECOTOXICOLÓGICA
Clasificación toxicológica y ecotoxicológica para el etiquetado del producto fitosanitario formulado y la Hoja de Datos de Seguridad, de acuerdo a la versión del Manual de las Naciones Unidas del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA), adoptada por la Autoridad Competente del Registro Nacional.
Se considerarán como excepción las clasificaciones de peligro de aves y abejas, las cuales adoptarán las clasificaciones de la USEPA.
XI - INFORMACIÓN SOBRE EL MANEJO DE DESECHOS DEL PRODUCTO FORMULADO
a.- Métodos de disposición final de los desechos Se debe incluir el o los procedimientos más adecuados para eliminación de remanentes o residuos de las aplicaciones, así como de los envases.
b.- Procedimientos para la destrucción del producto formulado y para la descontaminación.
Se deben presentar los procedimientos y métodos a seguir para la destrucción o inactivación del producto formulado.
Incineración controlada (condiciones).
Posibilidades de neutralización.
c.-Procedimientos de recuperación en caso de derrame Detallar el o los procedimientos para la recuperación del producto formulado.
d.- Depuración de las aguas Detallar el o los procedimientos a seguir para la depuración de fuentes de agua contaminadas con el producto formulado.
e.- En caso de incendio Identificar los productos de reacción y gases de combustión poniendo énfasis en aquellas sustancias que presentan riesgo toxicológico humano o ambiental y con base en lo anterior establecer y presentar un protocolo de respuesta ante una emergencia específica.
XII - EVALUACIÓN DE RIESGO
La Autoridad Competente establecerá qué categoría de productos deberán presentar una evaluación de riesgo en cumplimiento con lo establecido en el Anexo VII en el que se detalla el análisis de riesgos.
DECLARACIÓN DE NUEVOS ESTABLECIMIENTOS ELABORADORES DE PRODUCTO FORMULADO REGISTRADO
Se presentará una sola declaración de composición cuali-cuantitativa representativa para todas las plantas para las que se solicita el registro, si el producto a registrar no presenta diferencias entre las mismas, lo que deberá indicarse bajo declaración jurada.
A tal efecto, debe presentar para cada nuevo establecimiento:
1) Declaración jurada
Formulario de declaración jurada indicando la razón social y dirección real del establecimiento o planta a través de la plataforma SIGTrámites del Senasa o por aquellos medios que el Senasa disponga a tal fin, firmada por el Apoderado o Representante Legal.
* En virtud que por declaración jurada del responsable titular del registro, no existen diferencias significativas entre los productos obtenidos en los diferentes establecimientos o plantas de un mismo productos formulado, de observarse incumplimiento por acciones de control sobre el productos formulado de un establecimiento o planta, las medidas que la Autoridad Competente adopte serán aplicadas a todos los establecimientos o plantas declaradas para el registro involucrado.
2) Certificado de Origen del producto formulado.
El Certificado de Origen debe ser original y emitido por el fabricante y estar debidamente legalizado por las autoridades del país de origen.
Debe incluir:
• identificación de la sustancia activa.
• contenido mínimo declarado en % p/p.
• Denominación/razón social y domicilio real del establecimiento elaborador.
• (*)Nombre y dirección de la empresa que registrará el producto ante SENASA.
En caso de que no figure esta información se deberá remitir una nota emitida por el establecimiento elaborador donde conste la autorización para realizar el registro del producto en el país.
NUEVA MARCA COMERCIAL PARA UN PRODUCTO FORMULADO YA REGISTRADO
Si un registrante solicita un registro de un producto ya registrado por el mismo solamente se debe mencionar expediente aprobado.
Si el registro corresponde a otra persona física o jurídica, deberá presentarse la autorización de uso correspondiente.
I. Información legal y administrativa.
• Proyecto de marbete según normativa vigente.
• Hoja de Datos de Seguridad, de acuerdo a la versión del manual de las Naciones de las Naciones Unidas del “Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos”, adoptada por la Autoridad Competente del Registro Nacional de Terapéutica Vegetal..
Formulario II (condicionalmente requerido). Autorización a terceros de hacer uso del registro de un principio activo grado técnico para el registro de un producto formulado.
Requisitos y procedimientos DE COADYUVANTES DE APLICACIÓN.
I. Información legal y administrativa.
• Formulario de solicitud del producto formulado con aptitud COADYUVANTE,
• Proyecto de marbete según normativa vigente.
• Hoja de Datos de Seguridad, de acuerdo a la versión del manual de las Naciones Unidas del “Sistema Globalmente Armonizado de Etiquetado de Productos Químicos”, adoptada por la Autoridad Registro Nacional de Terapéutica Vegetal.
• Sistema de gestión de envases vacíos aplicado en de la Ley vigente.
II. Cuerpo técnico
1. Certificado de Origen del producto formulado.
El Certificado de Origen emitido por el fabricante (*) para cada uno de los orígenes a registrar, debe incluir:
• identificación de la sustancia principal que se pretende registrar como coadyuvante.
• contenido mínimo declarado.
• tipo de formulación
• Denominación/razón social y domicilio real del establecimiento fabricante.
(**)Nombre y dirección de la empresa que registrará el producto ante SENASA.
(*) Plantas del extranjero debidamente legalizado por las autoridades del país de origen.
(**) En caso de que no figure esta información se deberá remitir una nota emitida por el establecimiento fabricante donde conste la autorización para realizar el registro del producto en el país
2. Declaración de composición cuali-cuantitativa del producto formulado
La misma debe estar firmada por el establecimiento fabricante o por la empresa registrante con carácter de declaración jurada:
• Nombre químico según criterios internacionales (IUPAC o CAS) y número CAS (cuando esté disponible) de la/s sustancia/s activa/s y demás componentes.
• Contenido nominal de la(s) sustancia(s) a registrar con clase de uso coadyuvante, en base 100 % de pureza, expresado en porcentaje en masa (% p/p) para los formulados sólidos o gaseosos, y en porcentaje masa/volumen (% p/v) para los formulados líquidos.
• Contenido nominal, naturaleza química y función específica de cada uno de los componentes restantes incluidos en la formulación.
• Tipo de formulación armonizada de acuerdo con la Norma IRAM 12074.
• Método de análisis.
Se presentará una sola declaración de composición cuali-cuantitativa representativa para todas las plantas para las que se solicita el registro en la que debe figurar la razón social y dirección real de cada uno de los establecimientos fabricantes.
3. Información toxicológica y ecotoxicológica
El Dossier debe contener información científica respaldatoria con la caracterización toxicológica y/o ecotoxicológica para la clasificación de los productos formulados en función de la información disponibles para clasificar los peligros de los componentes principales y los co-formulantes, siendo posible aplicar los principios de extrapolación y estimación de la toxicidad aguda (ETA), según el manual de las Naciones Unidas del “Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos’’.
Requisitos y procedimiento DE SEMIOQUÍMICOS EN BASE A SUSTANCIAS ACTIVAS SINTÉTICAS.
Requisitos generales de registro de productos formulados que entre sus componentes contengan productos sintéticos, solos o mezcla con sustancias de origen biológico, que por su forma de uso se espera generen una respuesta en la conducta o en la fisiología de organismo plaga que se pretende controlar o monitorear. Comprenden a:
• Dispositivos de captura, cuando el uso del semioquímico tenga como objetivo la atracción sexual o alimentaria de plagas, con el objetivo de monitorear poblaciones de una especie.
• Atracticidas, cuando mediante el uso de cebos o de dispositivos de captura masivos, se combina el uso de semioquímicos (como atrayente sexual o alimentario), con el empleo de insecticidas o mecanismos de captura física para disminuir la abundancia y daños de las plagas en los cultivos.
• Confusión Sexual, cuando mediante el asperjado o el uso masivo de emisores de semioquímicos se busca la saturación del medioambiente con una feromona sexual con el fin de interferir en comportamiento de apareamiento de la plaga, para disminuir la abundancia y daños de las plagas en los cultivos.
• Repelentes, cuando mediante el asperjado o el uso masivo de emisores se busca la repelencia de plagas a fin de disminuir los daños de las mismas en los cultivos.
I. Información legal y administrativa.
• Formulario de solicitud del producto formulado con aptitud SEMIOQUÍMICO,
• Patrones: en caso de que la autoridad de aplicación así lo requiera, a los fines de control y vigilancia, la empresa registrante deberá aportar los patrones y metodología analítica de la/s sustancia/s activa/s, en las condiciones que establezca la Dirección General de Laboratorios y Control Técnico (DGLCyT) del SENASA.
• Proyecto de marbete según normativa vigente
• Hoja de Datos de Seguridad, de acuerdo a la versión del manual de las Naciones Unidas del “Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos’”, adoptada por la Autoridad Competente del Registro Nacional de Terapéutica Vegetal.
• Sistema de gestión de envases vacíos aplicado en cumplimiento de la Ley vigente, si corresponde.
II. INFORMACIÓN TÉCNICA.
NOTA: los productos formulados utilizados exclusivamente para Monitoreo de Plagas Agrícolas se encuentran exentos de presentación de información técnica respaldatoria
• Certificado de Origen de la Sustancia activa grado técnico.
El Certificado de Origen emitido por el fabricante (4), debe incluir:
• identificación de la sustancia activa.
• contenido mínimo declarado.
• Denominación/razón social y domicilio real del establecimiento fabricante.
(**) Nombre y dirección de la empresa que registrará el producto ante SENASA.
(*) Establecimiento del extranjero debidamente legalizado por las autoridades del país de origen.
(**) En caso de que no figure esta información se deberá remitir una nota emitida por el establecimiento fabricante donde conste la autorización para realizar el registro del producto en el país.
• Certificado de Origen del producto formulado.
El Certificado de Origen emitido por el fabricante (*) para cada uno de los orígenes a registrar, debe incluir:
• identificación de la/s sustancia/s activa/s.
• contenido mínimo de SUSTANCIA ACTIVA declarado.
• tipo de formulación
• Denominación/razón social y domicilio real del establecimiento fabricante.
• (**) Nombre y dirección de la empresa que registrará el producto ante SENASA.
(*) Establecimiento del extranjero debidamente legalizado por las autoridades del país de origen.
(**) En caso de que no figure esta información se deberá remitir una nota emitida por el establecimiento fabricante donde conste la autorización para realizar el registro del producto en el país.
• Declaración de composición cuali-cuantitativa del producto formulado
La misma debe estar firmada por el responsable del establecimiento fabricante o de la empresa registrante con carácter de declaración jurada:
• Nombre químico según criterios internacionales (IUPAC o CAS) y número CAS (cuando esté disponible) de la/s sustancia/s activa/s y demás componentes.
• Contenido nominal de la(s) sustancia(s) activa(s), en base 100 % de pureza, expresado en porcentaje en masa (% p/p) para los formulados sólidos o gaseosos, y en porcentaje masa/volumen (% p/v) para los formulados líquidos.
• Contenido nominal, naturaleza química y función específica de cada uno de los componentes restantes incluidos en la formulación.
• Tipo de formulación armonizada de acuerdo con la Norma IRAM 12074.
Se presentará una sola declaración de composición cuali-cuantitativa representativa para todas las plantas para las que se solicita el registro en la que debe figurar la razón social y dirección real de cada uno de los establecimientos fabricantes.
• Certificado de Análisis cuali-cuantitativo (condicionalmente requerido*)
Certificado de Análisis cuali-cuantitativo de una muestra representativa de UN (1) lote de formulación del producto formulado a registrar con nombre y firma del Responsable técnico del laboratorio que realizó el análisis.
• Propiedades físicas y químicas del producto formulado
Se requerirá la presentación de estudios para aquellos semioquímicos para los que se espera exposición (i.e. pulverizables), para los restantes se podrá presentar dossier sobre la base de la información técnica disponible para la/a sustancia/a activa/s y los coformulantes.
• Aspecto (estado físico y color):
• Densidad.
• pH.
• Estabilidad en el almacenamiento
• Tensión superficial (cuando corresponda)
• Solubilidad (cuando corresponda)
• Suspensibilidad (cuando corresponda)
• Ensayos de eficacia agronómica y fitotoxicidad (cuando corresponda).
En caso de no existir antecedentes de uso autorizado en Argentina, en relación a la naturaleza química del semioquímico (dosis, tipo de formulación/dispositivo o técnica de aplicación, etc), deben presentarse informes de los ensayos de eficacia agronómica, de acuerdo con lo establecido en el presente Manual según lo establecido en el Cap. 20 del presente manual.
• Información toxicológica y ecotoxicológica
Debe presentar un Dossier con información científica respaldatoria con la caracterización toxicológica para la clasificación de los productos formulados en función de la información disponible para clasificar los peligros de los ingredientes activos y los co-formulantes, siendo posible aplicar los principios de extrapolación y estimación de la toxicidad aguda (ETA), según el manual de las Naciones Unidas del “Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos”.
PRODUCTOS FORMULADOS A BASE DE AGENTES MICROBIANOS,
SUSTANCIAS BIOQUÍMICAS O INVERTEBRADOS
REQUISITOS GENERALES
a) Los Ensayos de eficacia agronómica deben ser realizados localmente, por especie plaga que se pretenda registrar, y cumplir con requisitos establecidos en la normativa vigente. Los ensayos de eficacia agronómica de un bioinsumo sin antecedentes de registro en el país, deben llevarse a cabo en al menos TRES (3) zonas agroecológicas distintas de la REPÚBLICA ARGENTINA, durante al menos DOS (2) campañas agrícolas. Podrán ser admitidos ensayos en una misma campaña siempre que la DAYB cerciore que los ensayos reflejen variabilidad climática, se realicen en al menos tres zonas agroecológicas y respeten el mínimo número de ensayos, SEIS (6).
b) Para bioinsumos a base de organismos modificados genéticamente, en todos casos deberá contar previamente al registro con la evaluación favorable realizada por la Coordinación de Innovación y Biotecnología perteneciente a la Dirección Nacional de Bioeconomía de SAGYP y la Comisión Nacional Asesora de Biotecnología Agropecuaria (CONABIA), conforme a lo establecido en las normativas vigentes. C) Cuando el producto a registrar proceda de la extracción de recursos natural nativo, la empresa registrante debe contar con el correspondiente permiso de colecta/acceso a los recursos genéticos.
INFORMACIÓN LEGAL Y ADMINISTRATIVA GENERAL.
Presentar de acuerdo con B - PRODUCTOS FORMULADOS - I. INFORMACION ADMINISTRATIVA.
Consideración para productos bioinsumos: Certificado de Origen firmado por personal responsable de la planta de producción / Establecimiento elaborador del Bioinsumo. En caso de provenir de una planta extranjera, deben estar debidamente autenticados y/o apostillados por las autoridades del país de origen.
INVERTEBRADOS PARA EL CONTROL BIOLÓGICO
I. Información legal y administrativa
• Acreditación del Registro de planta de cría.
• Certificado de origen del hospedero/presa que acompaña al invertebrado (cuando corresponda)
• Permiso de importación del invertebrado y del hospedero/presa que acompaña al invertebrado (cuando corresponda)
• Certificado de la Cuarentena emitido por el Laboratorio oficial (cuando corresponda)
• Autorización de colecta emitida por la autoridad de aplicación ambiental (cuando corresponda).
• Muestra de referencia para ser conservada en el laboratorio del SENASA.
II. Información técnica
A. Identidad y proceso de fabricación
• Nombre científico e identificación taxonómica (indique sinonimia, subespecie, variedad o raza si corresponde).
• Descripción morfológica.
• Método utilizado para identificar el invertebrado (taxonomía clásica, técnicas moleculares, etc.). Incluir claves de identificación morfológica cuando corresponda
• Estado de desarrollo biológico del invertebrado.
• Descripción detallada de los sustratos orgánicos e inorgánicos incluidos en el
• producto comercial.
• Nombre científico, características y función del hospedero/presa que acompaña al
• invertebrado (cuando corresponda).
• Libro de procedimiento y control de calidad para las etapas de crianza de los invertebrados.
B. Propiedades biológicas y utilización agronómica.
• Rango de hospederos/presas conocidos.
• Pruebas de especificidad, que indiquen cualquier riesgo potencial sobre especies no objetivos o para la salud de las personas y animales.
• Antecedentes de los resultados, de la introducción y/o liberación del organismo en otros países (para organismo exóticos).
• Estabilidad frente a diferentes condiciones climáticas
• Tipo de organismo (depredador, parasitoide o parásito), y estado o fase de desarrollo en el que será comercializado.
• Condiciones de liberación
• Método de liberación
• Dosis
• Intervalo entre liberaciones
• Periodo en que deben suspenderse las aplicaciones de otros productos fitosanitarios
• (antes y después de la liberación)
• Vida útil del producto
• Condiciones de almacenamiento, transporte y seguridad
• Informes de los ensayos de eficacia agronómica para los usos propuestos.
C. Envases y Embalajes según B - PRODUCTOS FORMULADOS - V. ENVASES Y EMBALAJES PROPUESTOS.
D. Medidas de Seguridad:
• Métodos recomendados y precauciones durante la multiplicación, fraccionamiento, almacenamiento, liberación y manipuleo general del producto. Indicar procedimientos de actuación en caso de accidentes.
Requisitos y procedimientos para SUSTANCIAS BIOQUIMICAS FORMULADAS A BASE DE EXTRACTOS DE ORIGEN VEGETAL, ANIMAL O MICROBIOLÓGICO.
I. Información legal y administrativa específica
a) Certificado de Composición original firmado por el Responsable Técnico de la empresa formuladora.
b) Informe de Análisis cuali-cuantitativo del producto formulado a registrar firmado por el Responsable técnico del laboratorio.
c) Autorización de colecta emitida por la autoridad de aplicación ambiental (cuando corresponda)
d) Acreditación del Registro de planta formuladora / establecimiento.
e) Sistema de gestión de envases vacíos aplicado en cumplimiento de la Ley vigente, si corresponde.
I. Información técnica
A. Información de la sustancia activa
a) Productor de la sustancia activa
b) Nombre común: Aceptado o propuesto por ISO y Sinónimo (si lo tiene).
c) Nomenclatura química (aceptado o propuesto por IUPAC), si corresponde.
d) Fórmula empírica, si corresponde.
e) Fórmula estructural (incluyendo estereoquímica de isómeros activos si corresponde), si corresponde.
f) Masa molecular, si corresponde.
g) Grupo químico al que pertenece.
h) Grado de pureza (contenido mínimo de sustancia activa en g/kg).
i) Caracterización de los otros componentes presentes en el extracto.
j) Caracterización de otros componentes agregados (ej. estabilizantes). Identificación y concentración.
B. Método de obtención de la sustancia activa
Esta información deberá venir respaldada por un Certificado emitido por el fabricante con el método de obtención, especificando los reactivos y solventes empleados en el proceso. Indicar los equipos utilizados y las condiciones de operación, especificando los parámetros controlados durante el proceso.
C. Métodos analíticos para la cuantificación y la identificación de la sustancia activa:
• El contenido mínimo declarado de la sustancia activa debe estar sustentado en el
• análisis de 5 lotes del grado técnico. Se deberá adjuntar la documentación de identificación y trazabilidad de los 5 lotes presentados
• Deberá presentarse el estudio completo de cuantificación de los 5 lotes, de acuerdo con los lineamientos de Buenas Prácticas de Laboratorio que defina la DGLyCT del Senasa, convenientemente firmado y fechado. El informe del laboratorio deberá incluir la descripción del método de ensayo, los registros cromatográficos, las tablas de cuantificación (incluyendo los datos crudos) y los Certificados de Análisis de los Patrones y la Muestra referidos en el estudio que deberán cumplir con protocolos internacionalmente reconocidos.
• Deberán facilitarse descripciones completas de la metodología que deberá estar convenientemente validada.
Deberán presentarse los parámetros de validación obtenidos:
• Linealidad.
• Límite de detección y cuantificación.
• Especificidad.
• Exactitud y repetibilidad.
Deberá realizarse la identificación de la sustancia activa en los 5 lotes por un método espectrométrico (MS, RMN, IR).
D. Propiedades físicas y químicas de la sustancia activa
a) Aspecto:
• Estado físico
• Color
b) Densidad
c) pH.
d) Estabilidad en el almacenamiento
B. Información del producto formulado
Identidad y proceso de fabricación del producto formulado
a) Declaración de la composición cuali-cuantitativa del producto formulado firmada por el Representante Legal con carácter de Declaración Jurada, que deberá incluir:
• Contenido de la(s) sustancia(s) activa(s), en base 100 % de pureza, expresado en porcentaje en masa (% p/p) para los formulados sólidos o gaseosos, y en porcentaje masa/volumen (% p/v) para los formulados líquidos.
• Contenido, naturaleza química y función específica de cada uno de los componentes restantes incluidos en la formulación.
• Tipo de formulación armonizada de acuerdo con la Norma IRAM 12074.
b) Análisis de una muestra representativa de UN (1) lote de formulación para la cuantificación de la(s) sustancia(s) activa(s). Deberá presentarse el estudio completo, de acuerdo con los lineamientos de Buenas Prácticas de Laboratorio que defina la DGLyCT del Senasa, convenientemente firmado y fechado,. El informe del laboratorio deberá incluir la descripción del método de ensayo, los registros cromatográficos, las tablas de cuantificación (incluyendo los datos crudos) y los Certificados de Análisis de los Patrones y la Muestra referidos en el estudio que deberán cumplir con protocolos internacionalmente reconocidos. Asimismo, se deberá adjuntar la documentación de identificación y trazabilidad de la muestra analizada.
Los valores de concentración informados para la(s) sustancia(s) activa(s) deberán cumplir con los límites establecidos en la Norma IRAM 12054, en función del contenido respectivo declarado.
c) Certificado de Origen del producto formulado, donde conste la identificación de la(s) sustancia(s) activa(s), su concentración declarada, el tipo de formulación armonizada y el nombre y la localización de la planta formuladora. El Certificado de Origen deberá estar debidamente legalizado por las autoridades del país de origen.
F. Descripción del proceso de formulación, que deberá contener la siguiente información:
a) Nombre y localización de la planta formuladora que interviene en el proceso.
b) Caracterización general del proceso: deberá consistir en una descripción completa y detallada de cada una de las etapas que constituyen el proceso, especificando qué componentes son agregados en cada una y qué operaciones se realizan.
c) Identificación de los ingredientes usados para formular el producto.
d) Descripción de los equipos usados, con sus especificaciones.
e) Descripción de las condiciones que se controlan en cada etapa del proceso y los parámetros de control de calidad del producto terminado.
f) Descripción de posibles reacciones posteriores al proceso de formulación que ocurran entre los ingredientes activos, o entre éstos y cualquier otro componente de la formulación o el envase; posible migración de materiales del envase al producto.
G. Propiedades físicas y químicas del producto formulado.
a) Aspecto:
• Estado físico.
• Color.
b) Densidad.
c) pH.
d) Estabilidad en el almacenamiento
e) Tensión superficial (cuando corresponda)
f) Suspensibilidad (cuando corresponda)
g) Solubilidad (cuando corresponda)
H. Información toxicológica, ecotoxicológica y ambiental
H.1. Toxicidad aguda en mamíferos.
a) Toxicidad oral aguda (DL50 oral)
b) Toxicidad cutánea aguda (DL50 cutánea)
c) Toxicidad aguda por inhalación (LC50 inhalación). Condicionalmente requerido
d) Irritación cutánea. Se deberá seguir un enfoque escalonado:
d.1. evaluación de la corrosividad dérmica utilizando un método de prueba in vitro validado;
d.2. un estudio de irritación dérmica inicial in vivo con un animal, y donde no se observan efectos adversos;
d.3. pruebas confirmatorias in vivo con uno o dos animales adicionales.
e) Irritación ocular. Se deberá seguir un enfoque escalonado:
e.1. el uso de una prueba de irritación / corrosión cutánea in vitro para predecir irritación / corrosión ocular;
e.2. un estudio inicial de irritación ocular in vivo con un animal, y donde no se observan efectos adversos;
e.3. pruebas confirmatorias in vivo con uno o dos animales adicionales.
f) Sensibilización cutánea.
Este estudio se requerirá a menos que el principio activo sea un sensibilizante conocido. Por ende, la no presentación de estudios de sensibilidad dermal debe ser documentada con un estudio puente donde se haya determinado que la sustancia es un sensibilizante dermal conocido.
g) Mutagenicidad:
Un estudio inicial de mutagenicidad es requerido mínimamente. Estudios subsiguientes pueden o no ser requeridos de acuerdo con los resultados obtenidos en los ensayos interiores.
Para cumplimentar este requisito se recomienda un enfoque por etapas: (IN VITRO)
g.1. Estudio de mutación genética en bacterias (por ej. Test de Ames)
g.2. Estudio de citogenética en células de mamíferos o test de micronúcleo. Este estudio no tiene que realizarse si hay datos negativos de estudios de citogenética in vivo.
g.3. Estudio de mutación genética en células de mamíferos, si se obtuvo un resultado negativo en g.1 y g.2., este estudio no será requerido.
g.4. Serán requeridos estudios de mutagenicidad in vivo en el caso de haber obtenido resultados positivos en los estudios in vitro.
H.2. Estudios subcrónicos y crónicos
Para nuevas sustancias sin antecedentes de registro en Argentina. Debe presentar un Dossier conteniendo información científica respaldatoria sobre la caracterización toxicológica, ecotoxicológica y destino ambiental. Esta información puede provenir de estudios toxicológicos y ecotoxicológicos realizados en laboratorio, mediante protocolos reconocidos internacionalmente, o de referencias bibliográficas completas procedentes de revistas indexadas. La evaluación de peligros se utilizará para cuantificar o desestimar los efectos potencialmente dañinos de la sustancia a ser comunicados en la etiqueta del producto y en la Hoja de datos de seguridad.
En aquellos casos, que por su forma de uso se espere exposición hacia el aplicador y el consumidor, debe presentar una Evaluación de la exposición y caracterización del riesgo dietario al consumidor, y una Evaluación de la exposición y caracterización del riesgo laboral
I. Ensayos de eficacia agronómica y fitotoxicidad.
Informes de los ensayos de eficacia agronómica de los usos propuestos.
J. Envases y embalajes propuestos según B -
PRODUCTOS FORMULADOS - V. ENVASES Y EMBALAJES PROPUESTOS.
AGENTES MICROBIANOS DE CONTROL BIOLÓGICO
Cada nuevo aislado se considerará como un nuevo ingrediente activo y debe registrarse independientemente de cualquier ingrediente activo registrado de manera similar. El nuevo aislamiento debe tener un identificador único después del nombre taxonómico del microorganismo. Esto no excluye la posibilidad de utilizar datos de otra cepa con antecedentes de registro, como estudios puente, para iniciar el proceso de registro.
Los productos elaborados en base a microorganismos sin antecedentes de registro en el país deberán realizar una solicitud de autorización de muestra y someterse a la evaluación de riesgo por parte autoridad competente en lo referente a los aspectos cuarentenarios y de impacto a nivel de los recursos naturales y/o de la salud. En caso de ser un microorganismo nativo, debe presentar la autorización de permiso de colecta de la provincia originaria.
Se considera como criterio excluyente de la presentación a registro los microorganismos que son patógenos primarios de animales, plantas y el ambiente. Aquellos que puedan presentar proximidad taxonómica con los microorganismos que son patógenos, deberán garantizar la inocuidad para la salud y el ambiente mediante los estudios que la avalen.
I. Información legal y administrativa
a) Certificado de Composición original con nombre y firmado por el Responsable Técnico de la empresa formuladora.
b) Informe de Análisis cuali-cuantitativo del producto formulado a registrar firmado por el Responsable técnico del laboratorio.
d) Acreditación del Registro de planta biológica / establecimiento elaborador.
e) Certificado de la Cuarentena emitido por el Laboratorio oficial (cuando corresponda)
II. Información técnica
A. Identidad y proceso de fabricación
a) Solicitante del registro.
b) Productor /proveedor de materia prima.
c) Nombre y descripción de la especie, caracterización de la cepa y origen. Taxonomía: la identificación deberá realizarse al menor nivel taxonómico en que se produzcan diferencias en el accionar del microorganismo o sus productos metabólicos (especie, subespecie, cepa, serotipo, patovar según corresponda al microorganismo), nivel de seguridad en la identificación por encima del 98%. Origen (nativo, exótico, OGM, etc).
d) Metodología de identificación y caracterización del microorganismo: Señalar la metodología y criterios utilizados para la identificación (morfología, bioquímica, serología, identificación molecular, etc.).
e) Datos de depósito de microorganismo: identidad de la colección y número de código de aislamiento.
f) Método de producción y control de calidad. Esta información deberá venir respaldada por una monografía, que contenga la descripción o flujograma del proceso productivo, especificando las condiciones, medios de cultivo, aditivos y otros empleados. Señalar las técnicas para garantizar la uniformidad y estandarización de la producción, y los procedimientos o metodología para el control de calidad.
g) Concentración del microorganismo (ingrediente activo) expresado en unidades infectivas conocidas. Declarar mínimo garantizado a la elaboración y al vencimiento, expresado en unidad de medida que corresponda.
h) Contenido e identidad de otros componentes. (Como condensados, medio de cultivo, promotores de la alimentación, entre otros). Señalar identidad y contenido máximo de microorganismos contaminantes, expresados en la unidad de medida que corresponda (CR).
i) Señalar identidad y caracterización de toxinas y metabolitos que forma el microorganismo, indicando, cuando corresponda, si tales toxinas o metabolitos ejercen o no la acción a la que está destinado al uso.
j) Estabilidad genética del agente de control biológico microbiano.
B. Métodos analíticos.
Indicar los métodos de análisis o técnicas, adjuntando su descripción, para:
a) Identificación del microorganismo al nivel establecido de especificidad (especie, subespecie, biotipo, cepa, etc.)
b) Determinación del contenido y viabilidad del microorganismo en el producto formulado.
C. Identidad del producto formulado.
a) Contenido del microorganismo: Indicar el contenido del o de los microorganismos en el producto formulado, indicando el contenido mínimo y nominal del material viable e inviable.
b) Co-formulantes: Indicar identidad, función y contenido máximo de aditivos, cuando corresponda; como los subproductos, condensados, medios de cultivo. Para la identificación de las sustancias químicas señalar nombre químico, nombre común, (si existe) y número CAS (si existe).
c) Certificado de origen y composición cuali-cuantitativo, emitido por el productor.
d) Propiedades físico químicas del producto formulado
• Color
• Estado físico
• pH
e) Estabilidad en diferentes condiciones ambientales (luz solar, pH, aire, temperatura, metales y sus iones).
f) Actividad acuosa (miscibilidad)
g) Estabilidad en el almacenamiento
h) Suspensibilidad (cuando corresponda)
C. Propiedades biológicas y utilización agronómica.
Informes de los ensayos de eficacia agronómica de los usos propuestos.
D. Evaluación tóxico-patológica. Efectos sobre la salud humana Debe presentar un Dossier con información científica respaldatoria con la caracterización toxico-patológica para la clasificación de los productos formulados en función de la información disponible para clasificar los peligros de los ingredientes activos y los co-formulantes. Mínimamente debe considerar información de toxicidad aguda/patogenicidad Oral aguda, Pulmonar aguda, Indicación de alergia/hipersensibilidad. Será condicionalmente exigido el cultivo de células (solo para virus). Para microorganismos sin antecedentes de uso en Argentina debe presentar información sobre la toxicidad/patogenicidad subcrónica
E. Evaluación tóxico-patológica. Efectos sobre otros organismos no objetivo
Debe presentar un Dossier con información científica respaldatoria con la caracterización ecotoxico-patológica para la clasificación de los productos formulados en función de la información disponible para clasificar los peligros de los ingredientes activos y los coformulantes en aves, peces de agua dulce y abejas.
F. Información respecto a la seguridad
Procedimientos para la destrucción del agente microbiológico, producto de su metabolismo, producto formulado, agentes biológicos mutantes, indicando las condiciones físicas o químicas específicas para obtener la desactivación o descomposición del material biológico/producto.
Métodos recomendados y precauciones de manejo durante la fabricación, formulación, almacenamiento, transporte, uso y manipuleo general del agente / producto. Información sobre equipos de protección personal (si corresponde). Procedimientos de limpieza y descontaminación de equipos de aplicación y áreas contaminadas.
G. Envases y embalajes propuestos según B - PRODUCTOS FORMULADOS - V. ENVASES Y EMBALAJES PROPUESTOS.
Consideraciones para preservadores de la madera: Los biocidas gaseosos para la fumigación de viviendas y artículos de madera como así de los destinados a reimpregnación de postes y los destinados a usos domisanitarios quedan excluidos de la obligatoriedad de inscribirse en el Registro Nacional.
PROCEDIMIENTO PARA LA EXTENSIÓN DEL VENCIMIENTO DE UN LOTE DE PRODUCTO FITOSANITARIO
Para los productos fitosanitarios vencidos o próximos a vencer, se podrá solicitar la extensión de la fecha de vencimiento de un lote dado de producto según el siguiente procedimiento:
1) Mediante la presentación de una solicitud, la firma registrante deberá identificar claramente el producto y el/los lote/s respectivo/s próximo/s a vencer o vencido/s.
2) SENASA procederá a realizar la toma de la/s muestra/s; O gestionar las mismas con la firma titular del producto.
3) La firma remitirá la/s muestra/s tomada/s a un laboratorio reconocido por SENASA para realizar los análisis pertinentes para comprobar que el lote del producto fitosanitario vencido o próximo a vencer se halla en condiciones para la extensión de su vida útil (cumple con la especificación).
4) Si corresponde extender la vida útil por un (1) año desde la fecha de vencimiento del lote, deberán obtenerse resultados conformes para la cuantificación de la/s sustancia/s activa/s de acuerdo con los límites establecidos en la Norma IRAM 12054.
5) Si corresponde extender la vida útil por dos (2) años desde la fecha de vencimiento del lote, deberán obtenerse resultados conformes para la cuantificación de la/s sustancia/s activa/s de acuerdo con los límites establecidos en la Norma IRAM 12054, y para el ensayo de estabilidad acelerada en almacenamiento.
6) La firma remitirá a SENASA el informe de los análisis efectuados junto con una nota, especificando: a. Marca comercial del producto b. Número de registro c. Lote d. Cantidad de unidades e. Tipo de presentación
7) En caso de poder extenderse la vida útil del lote en cuestión, el registrante deberá modificar la fecha de vencimiento (el resto de la información deberá mantenerse). Esto implica ubicar el nuevo vencimiento en el envase primario (y en el empaque secundario, si es que el producto lleva su vencimiento en la caja).
8) La firma registrante informará al Sistema Nacional de Trazabilidad la nueva fecha de vencimiento para el lote en cuestión, si así correspondiera.
REQUISITOS Y PROCEDIMIENTOS PARA PRODUCTOS DE TRATO DIFERENCIADO (PTD).
Se considera Producto de Trato Diferenciado a todo aquel que por sus características físicas y químicas, toxicológicas, ecotoxicológicas, modo de uso u otra cuestión técnica, no es alcanzado por la totalidad de los requisitos, o por su naturaleza o estado de avance técnico al momento de la presentación de la solicitud de registro.
Las personas físicas o jurídicas que inician un procedimiento de registro, podrán solicitar el tratamiento diferenciado de un principio activo o producto formulado, si corresponde, mediante la presentación de la información y documentación que fundamente dicho trato (Waiver), como anexo a cada tipo de categoría solicitada, a ser:
SUSTANCIAS ACTIVAS GRADO TÉCNICO NUEVAS.
SUSTANCIAS ACTIVAS GRADO TÉCNICO EQUIVALENTES.
PRODUCTOS FORMULADOS EN BASE A SUSTANCIAS ACTIVAS GRADO TÉCNICO NUEVAS O EQUIVALENTES.
COADYUVANTES DE APLICACIÓN, SEMIOQUÍMICOS Y FORMULADOS MISCELÁNEOS.
AUTORIZACIÓN DE PREDIOS EXPERIMENTALES Y DE USO EXPERIMENTAL DE SUSTANCIAS ACTIVAS SINTÉTICAS.
PRODUCTOS FORMULADOS EN BASE A SUSTANCIAS ACTIVAS BIOQUÍMICAS. PRODUCTOS FORMULADOS MICROBIANOS DESTINADOS A LA PROTECCIÓN VEGETAL
FISCALIZACIÓN
Fiscalización post autorización de la sustancia activa grado técnico nueva o equivalente o del producto formulado. El SENASA podrá efectuar fiscalizaciones post autorización de la sustancia activa grado técnico o del producto formulado mediante análisis de muestras del producto autorizado, estando el costo de envío de muestras y análisis a cargo del titular del registro o del importador. En caso de que la fiscalización demuestre inconsistencias entre la información declarada y los análisis efectuados sobre las muestras oficiales, el mencionado Servicio Nacional dará de baja la autorización de conformidad con los procedimientos establecidos en la normativa vigente, sin perjuicio de las sanciones que pudieran corresponder de acuerdo con lo dispuesto en el Capítulo V de la Ley N° 27.233 y su Decreto Reglamentario N° DECTO-2019-776-APN-PTE del 19 de noviembre de 2019, y la eventual aplicación de las medidas preventivas pertinentes.
Fiscalización documental post autorización de la sustancia activa grado técnico y/o del producto formulado. El SENASA podrá efectuar fiscalizaciones documentales post autorización de la sustancia activa grado técnico y/o del producto formulado. En caso de que la fiscalización demuestre inconsistencias entre la información declarada y la presentada al solicitar la autorización de la sustancia activa o del formulado para la planta en cuestión, el citado Servicio Nacional dependiendo del grado de inconsistencia dará de baja la autorización de conformidad con los procedimientos establecidos en la normativa vigente o solicitará la subsanación correspondiente, sin perjuicio de las sanciones que pudieran corresponder de conformidad con lo establecido en el Capítulo V de la mentada Ley N° 27.233 y su referido decreto reglamentario.
Las muestras oficiales de fiscalización y control serán remitidas a laboratorios que cuenten, al menos, con certificación de Buenas Prácticas de Laboratorio (BPL), es decir, que cumplan con las mismas exigencias de laboratorios que realicen estudios y análisis con objeto de evaluación para solicitud de registro.
En caso de detectarse alguna irregularidad relacionada con la calidad del producto o con la documentación declarada en productos con certificaciones de registro emitidas por las autoridades competentes de los países o grupos de países individualizados en el Anexo IV (IF-2025-68648006-APN-DNPV#SENASA), la autoridad competente podrá adoptará serán las siguientes acciones:
a) Suspender preventivamente el registro.
b) Dependiendo de la gravedad de lo hallado, se podrá solicitar la subsanación de la documentación presentada (bajo régimen de caducidad y pausado) y en caso de no ser posible la subsanación se procederá a la cancelación del registro.
c) dar de baja en el fitosanitarios en el VADEMECUM de acuerdo con la evaluación de las subsanaciones o cancelación.
d) De reiterarse TRES (3) irregularidades en las DDJJ del mismo o de distintos productos de una determinada firma registrantes, la Clave Única de Identificación Tributaria (C.U.I.T.) de la firma involucrada ingresará al régimen de vigilancia intensiva de monitoreo durante un año y dependiendo de la gravedad del incumplimiento se procederá a restringir por SEIS (6) meses la posibilidad de realizar cualquier actividad vinculada al comercio de fitosanitarios,
e) Estas medidas son tomadas sin perjuicio de aquellas otras medidas legales que pudieran adoptarse de acuerdo con la gravedad o el tipo de falta y las competencias intervinientes que correspondan.
SUSTANCIAS ACTIVAS GRADO TÉCNICO NUEVAS, EQUIVALENTES Y/O PRODUCTOS FORMULADOS.
Cuando los productos fitosanitarios cuenten con la aprobación de uso agrícola, por las Autoridades Competentes del listado de países o grupos de países que se detallan en el Anexo IV de la presente norma, deberán cumplir con lo prescripto en su Artículo 7°, según el procedimiento que a continuación se detalla:
I. INFORMACIÓN ADMINISTRATIVA
1) Formulario con carácter de declaración jurada, informando el Grado Técnico Nuevo o Equivalente y/o productos formulados a través de la plataforma SIG-Trámites del SERVICIO NACIONAL DE SANIDAD Y CALIDAD AGROALIMENTARIA (SENASA) o la que en el futuro la reemplace, firmada por el Apoderado o Representante Legal y el Responsable Técnico con incumbencia profesional en la materia.
DATOS DEL SOLICITANTE:
Nombre y Apellido;
Razón Social;
Dirección;
Teléfono;
Correo Electrónico;
DATOS DEL PRODUCTO A IMPORTAR:
Nombre químico;
Número de código experimental;
Grupo químico;
Concentración de Sustancia Activa;
Características/Estado Físico;
Establecimiento productor,
País de origen de la sustancia;
2) Presentar el documento oficial de registro y/o comercialización emitido por la Autoridad Competente que integre el listado de países mencionado en el Anexo IV de la presente norma, donde conste la denominación de la sustancia activa grado técnico, la pureza mínima expresada en PORCENTAJE PESO EN PESO (% P/P), el nombre y el domicilio del establecimiento sintetizador.
3) Hoja de Datos de Seguridad, de acuerdo con la versión del Manual de las Naciones Unidas del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA), adoptada por la Autoridad Competente del Registro Nacional.
En el caso de que los productos NO se comercialicen en la REPÚBLICA ARGENTINA, se deberá declarar además de lo solicitado en los Puntos 1, 2, y 3, lo siguiente:
- el límite máximo de residuos (LMR) establecido en el país en el que el producto se encuentra registrado, indicando las condiciones bajo las cuales dicho límite fue determinado;
- el LMR establecido en el país en el que el producto se encuentra registrado para el o los cultivos a tratar, y el límite de cuantificación analítica (LOQ);
- el cultivo objetivo al que está destinado;
- organismo nocivo o plaga a ser controlada,
- la dosis de aplicación recomendada,
- período de carencia para cada cultivo,
- Registro Nacional Sanitario de Productores Agropecuarios (RENSPA) donde será utilizado.
La finalidad de esta declaración jurada es demostrar que el producto fitosanitario no representa un riesgo para la salud humana, animal ni para el ambiente.
Alternativamente, el solicitante podrá optar por presentar los resultados de ensayos de eficacia agronómica y LMR realizados en la REPÚBLICA ARGENTINA, a fin de acreditar que el producto no incurre en riesgos para la salud humana, animal ni ambiental en las condiciones locales de uso.
II. INFORMACIÓN Y DOCUMENTACIÓN CONFIDENCIAL.
Información confidencial:
- Declaración de pureza e impurezas para sustancias activas.
- Declaración de composición en los formulados.
La información y documentación confidencial será reservada al sólo efecto de la fiscalización, y para el caso de sustancias activas nuevas, como referencia de sustancias activas equivalentes.


AVISO DE IMPORTACIÓN DE MUESTRAS DE PRODUCTOS FITOSANITARIOS EN ETAPAS TEMPRANAS DE DESARROLLO
Para cada sustancia o producto de la que se desee importar una muestra, se deberá presentar un aviso de importación con carácter de declaración jurada, la cual será automática, a través de la Plataforma SIG-Trámites o la que en el futuro la reemplace, que contenga la siguiente información:
Datos del solicitante:
1. Nombre;
2. Dirección;
3. Teléfono;
4. Correo Electrónico;
5. Responsable legal
1. Datos del producto a importar:
1. Nombre químico;
2. Número de código experimental;
3. Grupo químico;
4. Concentración de Sustancia activa;
5. Características/estado físico;
6. Establecimiento productor, país de origen de la sustancia;
7. Destino dentro del país y/o localidad en que se realizará la prueba (Predio de experimentación);
8. Cantidad de sustancia, en unidades y/o kilogramos y/o litros;
9. Justificación de la cantidad de muestra necesaria para las pruebas en ejecución y/o en proyecto,
10. Hoja de Datos de Seguridad, de acuerdo a la versión del manual de las Naciones de las Naciones Unidas del “Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos”, adoptada por la Autoridad Competente del Registro Nacional de Terapéutica Vegetal.
Las muestras en etapa de desarrollo no podrán ser comercializadas, dado que estas están destinadas exclusivamente a uso experimental. Su ingreso al país se realizará bajo la entera responsabilidad de quien las introduzca, debiendo garantizar el cumplimiento de las normativas vigentes y las condiciones establecidas para su manipulación y uso.
Las muestras deberán tener en su identificación adherida al envase la frase: 'MUESTRA SIN VALOR COMERCIAL, PROHIBIDA SU VENTA'.

Después de la aprobación de un componente activo o del registro de un producto fitosanitario, puede haberse generado nueva información científica que sugiera la existencia de riesgos para la salud humana, la seguridad animal o el medio ambiente previamente desconocidos, así como también en el comercio de productos agrícolas o incluso que surgieran datos e información asociados a problemas o disminución de la eficacia agronómica de un producto fitosanitario.
El proceso de reevaluación o revisión de productos fitosanitarios registrados tiene como propósito que, mediante la herramienta de análisis de riesgo, la Autoridad Competente determine si en función de nuevos antecedentes técnicos y científicos respecto a la caracterización de peligros, evaluación de la exposición, cambios en la eficacia de los productos autorizados debido a cambios tecnológicos o aparición de resistencia de plagas, entre otras cuestiones, resulta procedente iniciar los procedimientos para modificar prácticas agrícolas, períodos de carencia, límites máximos de residuos, vigencia de las autorizaciones, realizar reclasificaciones, establecer restricciones parciales o totales de un producto fitosanitario o cualquier otra medida preventiva o correctiva cuando los usos autorizados vigentes de ese producto puedan causar efectos adversos, en las condiciones locales de uso, en niveles inaceptables tanto para la salud como para el ambiente o pueda verse afectada la eficacia agronómica en las condiciones locales de dicho producto o afecte el comercio de los productos agrícolas o alimentos que se comercialicen.
De este modo, la revisión de registros tiene como objetivo garantizar que, a medida que evoluciona la capacidad de evaluar riesgos y cambian las caracterizaciones de las sustancias y las prácticas, todos los productos fitosanitarios registrados sigan cumpliendo con el estándar legal de ausencia de efectos adversos inaceptables a la salud y el ambiente.
Sin perjuicio de la aplicación del proceso de reevaluación o revisión sobre productos fitosanitarios por las razones indicadas, la Autoridad Competente puede tomar las medidas preventivas o correctivas que correspondan sin que medie revisión, si las razones que ameritan tales medidas se fundamentan en cuestiones comerciales o situaciones de emergencia que requieran de ellas, pudiendo abstraerse de la reevaluación o revisión.
La Autoridad Nacional Competente del Registro Nacional podrá realizar un plan de priorización plurianual para la reevaluación de usos locales de sustancias activas relacionadas con posibles cambios en el perfil de riesgo en la i) Exposición alimentaria; ii) Exposición laboral; iii) Exposición ambiental; y iv) Eficacia agronómica/resistencia de plagas, así como también debido al surgimiento de nueva información disponible sobre los perfiles toxicológicos, ecotoxicológicos y el destino ambiental de las sustancias activas.
El proceso formal de reevaluación o revisión de productos fitosanitarios registrados se inicia cuando la Autoridad Competente envía una notificación a los registrantes, o publica oficialmente una decisión técnica debidamente justificada, informando su decisión de someter las autorizaciones de uso aprobadas, y/o registros, a revisión.
El cumplimiento de este aviso es un requisito legal para los titulares que deseen mantener las autorización de uso vigente, y se aplicarán sanciones por no proporcionar la información requerida dentro del plazo especificado en el plan de trabajo propuesto. El plan de trabajo para la reconsideración será discutido con los titulares de registro y se establecerán hitos clave y el plazo normativo dentro del cual se debe tomar una decisión sobre las condiciones de autorización de usos en evaluación.
Además de los datos presentados por los titulares de registro, la autoridad competente podrá considerar todos los datos publicados e informes de evaluación relevantes recabados por otras agencias regulatorias.
Los estudios requeridos que surgieran del plan de trabajo serán responsabilidad compartida por todas las empresas afectadas y/o empresas interesadas en participar en conjunto, siendo la distribución de tareas y costos responsabilidad de las propias empresas, debiendo estas proceder a su distribución en base a los costos efectivos de los estudios realizados y las participaciones respectivas en el mercado local.
Todas las empresas afectadas deben aceptar las decisiones de reevaluación o revisión de la Autoridad Competente, las cuales pueden estar fundadas en los motivos enumerados a continuación:
- Adecuación de límites máximos de residuos y tiempos de carencia.
- Cancelación de usos.
- Adecuación de dosis y/o momentos de aplicación.
- Modificación del espectro de control de plagas informado previamente.
- Restricción de formas de aplicación.
- Reclasificación de peligro/riesgo.
- Cancelación de determinadas formulaciones.
- Cancelación de registros.
- Toda otra modificación, restricción o cancelación, no contemplada expresamente.
Pueden recibirse solicitudes de registro de sustancias activas y sus productos formulados puestos en reevaluación o revisión debido a nuevos antecedentes técnicos y científicos respecto a la caracterización de peligros toxicológicos o ecotoxicológicos, a cambios en la eficacia o aparición o existencia confirmada de resistencia de plagas.
No se aceptarán nuevos usos o modificaciones de recomendaciones de uso de las sustancias en revisión mientras se encuentre activa la reevaluación y hasta tanto la Autoridad Competente se expida sobre la decisión a adoptarse como resultado de la revisión.
Las personas humanas o jurídicas que presenten solicitudes de registro de productos fitosanitarios que se encuentren en reevaluación deben declarar su conocimiento del plan de trabajo comunicado y de los hitos clave y los plazos establecidos, así como también el compromiso de su cumplimiento.

CLASIFICACIÓN, COMUNICACIÓN DE LOS PELIGROS PARA LA SALUD, Y PROTOCOLO DE ENSAYOS DE EFICACIA AGRONÓMICA, FITOTOXICIDAD Y RESIDUOS
PROTOCOLO DE ENSAYOS DE EFICACIA AGRONÓMICA Y FITOTOXICIDAD
PROPÓSITO Y ALCANCES:
El objetivo de los ensayos de eficacia agronómica es proporcionar resultados comparables y confiables sobre la eficacia de un producto fitosanitario a registrar o de su recomendación de uso, considerando las condiciones climáticas y agronómicas locales, de acuerdo con las Buenas Prácticas Agrícolas (BPA) propuestas en el marbete del producto fitosanitario. Los resultados de los ensayos de eficacia y fitotoxicidad son la base para el establecimiento de la Buena Práctica Agrícola Crítica que se utilizará para la definición del LMR para el uso propuesto.
REQUISITOS DE PRESENTACIÓN:
La Autoridad Competente deberá solicitar ensayos de eficacia agronómica para aquellos productos fitosanitarios registrados o en vías de registro, cuando deba reunir evidencias significativas para:
- Cuantificar la eficacia agronómica de un producto formulado a registrar con base en sustancias activas grado técnico nuevas.
- Cuantificar la eficacia agronómica de un producto formulado registrado con base en sustancias activas grado técnico equivalentes, en un nuevo cultivo.
- Cuantificar la eficacia agronómica de un producto formulado registrado con base en sustancias activas grado técnico equivalentes, para el control de una nueva plaga.
- Cuantificar la eficacia agronómica de un producto formulado registrado con base en sustancias activas grado técnico equivalentes, ante un cambio de dosis o momento de aplicación.
- Cuantificar la eficacia agronómica de una nueva combinación de principios activos o el cambio en la proporción de sustancias activas grado técnico equivalentes, en un producto formulado en mezcla.
PROCESO DE VERIFICACIÓN:
La empresa registrante debe notificar semestralmente ante la Autoridad Competente, la planificación semestral de ensayos de eficacia agronómica. En dicha planificación debe indicar los sitios donde se realizarán los ensayos de eficacia agronómica, la fecha estimada de inicio y de las evaluaciones postaplicación y los datos de contacto del ensayista responsable, a fin de que la autoridad regulatoria pueda realizar el seguimiento y solicitar información adicional cuando lo considere necesario con fines de verificación.
Los ensayos podrán ser constatados o verificados por la Autoridad Competente en aquellos puntos que considere críticos del proceso del ensayo de eficacia agronómica (a saber: dosificación, aplicación, monitoreo u otro), por lo que cada una de estas actividades deben estar debidamente registradas en los medios físicos y/o digitales que el ensayista responsable considere adecuados.
REQUISITOS MÍNIMOS DE PLANIFICACIÓN DE ENSAYOS LOCALES:
Los ensayos de eficacia agronómica deberán llevarse a cabo donde el cultivo en estudio es realizado comercialmente, con niveles apropiados de infecciones o infestaciones de plagas que permitan verificar la eficacia de los tratamientos evaluados. Deben reflejar las recomendaciones de uso que se deberán incorporar en la etiqueta del producto.
Al menos SEIS (6) ensayos de eficacia agronómica deberán llevarse a cabo en diferentes zonas agroecológicas de la REPÚBLICA ARGENTINA, representativas de la combinación plaga-cultivo para la que solicita la ampliación de uso. Los ensayos deberán realizarse durante al menos DOS (2) campañas agrícolas.
En casos excepcionales, se podrán aceptar protocolos de ensayos de eficacia y fitotoxicidad en una campaña agrícola, previa justificación técnica a la Autoridad Competente que motive el trato diferenciado. En tales casos, los lotes experimentales ubicados en una misma región agroecológica, deben estar distanciados al menos VEINTE KILÓMETROS (20 km) entre sí.
Para cultivos considerados menores en la REPÚBLICA ARGENTINA, se requerirán al menos DOS (2) ensayos locales.
El plan de ensayos de estudios de residuos locales puede desarrollarse considerando tres categorías de cultivos:
- Cultivos mayores, representativos en Argentina.
- Cultivos menores.
- Cultivos mayores, NO representativos en Argentina.
ESTABLECIMIENTO DE RESIDUO POR ESPECIE VEGETAL, EN CULTIVOS MAYORES REPRESENTATIVOS EN ARGENTINA:
Se requerirán al menos SEIS (6) ensayos de residuos, de los cuales, al menos CUATRO (4) deberán ser locales, realizados en áreas agroecológicas diferentes y representativas de cada cultivo, en la misma temporada o en temporadas diferentes (ver Tabla Agrupación de Cultivos, mayores representativos y de cultivos menores para la REPÚBLICA ARGENTINA). Optativamente, serán aceptados hasta DOS (2) estudios de residuos realizados bajo Buenas Prácticas de Laboratorio (BPL) conducidos en otros países, siempre que la recomendación de uso del ensayo sea comparable con la práctica agrícola crítica a registrar.
ESTABLECIMIENTO DE RESIDUO POR GRUPOS DE CULTIVO, SOBRE LA BASE DE CULTIVOS MAYORES REPRESENTATIVOS EN ARGENTINA:
Se requerirán al menos OCHO (8) ensayos de residuos locales, en especies representativas pertenecientes a los grupos de cultivos definidos en el presente Anexo (ver Tabla Agrupación de Cultivos, mayores representativos y de cultivos menores para la REPÚBLICA ARGENTINA), en áreas agroecológicas diferentes y representativas de cada cultivo, en la misma temporada o en temporadas diferentes. Optativamente, serán aceptados hasta el VEINTICINCO POR CIENTO (25 %) del número total de estudios de residuos BPL, que hayan sido conducidos en otros países, siempre que la recomendación de uso del ensayo sea comparable con la práctica agrícola crítica a registrar.
ESTABLECIMIENTO DE RESIDUO POR GRUPOS DE CULTIVO Y POR ESPECIE VEGETAL EN CULTIVOS MAYORES NO REPRESENTATIVOS EN ARGENTINA:
Se requerirán al menos SEIS (6) ensayos de residuos BPL para fijación de tolerancia por grupos, y CUATRO (4) para fijación de tolerancias por especie mayor NO representativa de Argentina, los cuales pueden haber sido conducidos en otros países, siempre que la recomendación de uso del ensayo sea comparable con la práctica agrícola crítica a registrar (ver Tabla Agrupación de Cultivos, mayores NO representativos y de cultivos menores para la REPÚBLICA ARGENTINA).
ESTABLECIMIENTO DE RESIDUO POR ESPECIE VEGETAL, EN CULTIVOS MENORES EN ARGENTINA:
Se requerirán al menos DOS (2) ensayos de residuos BPL conducidos en Argentina o en otros países, siempre que la recomendación de uso del ensayo sea comparable con la práctica agrícola crítica a registrar (ver Tabla Agrupación de Cultivos, mayores representativos y de cultivos menores para la REPÚBLICA ARGENTINA y Tabla Agrupación de Cultivos, mayores NO representativos y de cultivos menores para la REPÚBLICA ARGENTINA);
Se considerarán excepciones a estos requisitos:
- Establecimiento de residuos para uso en post-cosecha:
Se requerirán al menos CUATRO (4) ensayos de residuos por especie vegetal o en cultivos representativos del correspondiente grupo de cultivos según lo establecido en las Tabla Agrupación de Cultivos, mayores representativos y de cultivos menores para la REPÚBLICA ARGENTINA y Tabla Agrupación de Cultivos, mayores NO representativos y de cultivos menores para la REPÚBLICA ARGENTINA. Estos ensayos pueden ser desarrollados localmente o conducidos en otros países, en este último caso, siempre que la recomendación de uso del ensayo sea comparable con la práctica agrícola crítica a registrar en Argentina.
- Aplicación en pre-siembra o en preemergencia del cultivo y aplicación de herbicidas entre las líneas de cultivos perennes en producción, aplicación en dormancia de cultivos perennes o cuando se cuente con ensayos presentados ante Autoridades regulatorias, hechos bajo BPL y que cumplan con la práctica crítica (BPA) a registrar, que demuestren que los residuos se encontrarán por debajo del límite de cuantificación:
Se requerirán DOS (2) ensayos de campo en regiones agroecológicas diferentes y representativas del cultivo, en la misma estación o en temporadas diferentes en el mismo lugar, si el resultado de la los valores de residuos son menores o iguales a un Límite de Quantificación (LOQ) de CERO COMA CERO UNA PARTES POR MILLÓN (0,01 ppm).
Sin embargo, si el resultado del estudio de residuo es mayor que el valor del LOQ, en al menos UNA (1) de las pruebas, se deberán realizar ensayos adicionales que cumplan con los criterios detallados en los puntos “Establecimiento de residuo por especie vegetal, en cultivos mayores representativos en Argentina” y “Establecimiento de residuo por grupos de cultivo, sobre la base de cultivos mayores representativos en Argentina”.
RESTRICCIONES DE USO DE MATERIAL TRATADO:
Los productos agrícolas y los restos de cultivo, provenientes de las áreas tratadas con los productos formulados a evaluar, no podrán ser utilizados para alimentación humana o animal. Cuando se trate de un ensayo de experimentación de productos utilizados en forma de trampas o cebos, que por su forma de uso no entren en contacto con el cultivo, podrá permitirse el consumo de los productos agrícolas para fines de alimentación humana o animal.
ANÁLISIS DE RESULTADOS:
Se deben presentar los resultados obtenidos tras la aplicación de diversas técnicas estadísticas a los datos.
En el texto del informe deben figurar solo los datos estadísticos principales, la información sobre la media y los errores estándar de las variables cuantificadas y comparadas entre los tratamientos evaluados.
AMPLIACIONES DE USO DE OFICIO PARA CULTIVOS MENORES O USOS MENORES
La Autoridad Competente puede, sobre la base de información suficiente y científicamente válida desarrollada en el país o en el exterior, establecer prácticas agrícolas y límites máximos de residuos de oficio en forma temporal o definitiva, dependiendo de la calidad de la información que disponga.
A tal fin, puede conformar comisiones o grupos de trabajo con la participación del INSTITUTO NACIONAL DE TECNOLOGÍA AGROPECUARIA (INTA), Universidades, expertos y especialistas en la materia, representantes de las áreas competentes en materia de productos fitosanitarios de los gobiernos provinciales, representantes de productores, organizaciones o instituciones de investigación y otras que con incumbencia en la materia colaboren en el desarrollo y autorización de usos en cultivos menores o usos menores con sustento científico.
CONCLUSIONES:
Pueden brindarse análisis comparativos de los resultados de esta investigación con los de otros autores, para evidenciar la contribución neta realizada por los ensayos, la corroboración de otros estudios, las discrepancias con ellos, etcétera. Cualquier información que respalde específicamente las declaraciones de etiqueta debe ser discutida, incluidos los efectos secundarios, si los hubiera.
Todos los ensayos que realicen los interesados y la información que surja de ellos sobre composición y procesos de elaboración serán reservados. Su vista queda reservada a personal y auxiliares de la Autoridad Competente afectada al proceso aprobatorio del producto, a los responsables técnicos designados y a las personas fehacientemente autorizadas por la firma que solicita la inscripción.
NOTA ACLARATORIA
Las tablas detalladas a continuación son a modo de guía conceptual, complementa, pero no reemplaza los criterios definidos en la Versión ST/SG/AC.10/30/Rev.9 del Manual de las Naciones Unidas del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA).
LAS TABLAS NO SE AGREGAN YA QUE NO TUVIERON COMENTARIOS.
NOTA ACLARATORIA
Las tablas detalladas a continuación son a modo de guía conceptual, complementa, pero no reemplaza los criterios definidos en la Versión ST/SG/AC.10/30/Rev.9 del Manual de las Naciones Unidas del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA).

Fuente: FAO and WHO. 2022. Guidance on good labelling practice for pesticides (Second revision)
- International Code of Conduct on Pesticide Management. Rome.
Criterios que definen las categorías de peligro de toxicidad aguda

Notas a la tabla:
a) La estimación de la toxicidad aguda (ETA) para la clasificación de una sustancia se deducirá a partir de la DL50/CL50, cuando se conozca;
b) Los valores de toxicidad aguda se expresan en valores (aproximados) de la DL50 (ingestión, absorción cutánea) o CL50 (inhalación) o en estimaciones de la toxicidad aguda (ETA).
c) Los valores de corte/límites de concentración para la toxicidad por inhalación que figuran en la tabla se basan en una exposición de CUATRO (4) horas. Para convertir los datos de modo que respondan a una exposición de UNA (1) hora, hay que dividirlos por DOS (2) para gases y vapores y por CUATRO (4) para polvos y nieblas;
d) Algunas reglamentaciones utilizan la concentración de vapor saturado como un elemento adicional para protecciones específicas de salud y seguridad (por ejemplo, la Reglamentación Modelo de las Naciones Unidas);
e) En algunas sustancias, la atmósfera del ensayo no será solo un vapor sino que consistirá en una mezcla de fases líquidas y gaseosas. En otras sustancias, esa atmósfera podrá consistir en un vapor próximo al estado gaseoso. En estos últimos casos, la clasificación [en partes por millón de volumen (ppmV)] será la siguiente: Categoría 1, CIEN PARTES POR MILLÓN DE VOLUMEN (100 ppmV); Categoría 2, QUINIENTAS PARTES POR MILLÓN DE VOLUMEN (500 ppmV); Categoría 3, DOS MIL QUINIENTAS PARTES POR MILLÓN DE VOLUMEN (2.500 ppmV); Categoría 4, VEINTE MIL PARTES POR MILLÓN DE VOLUMEN (20.000 ppmV). Los términos “polvo”, “niebla” y “vapor” se definen como sigue:
- Polvo: partículas de una sustancia o de una mezcla en suspensión en un gas (en el aire por lo general)
- Niebla: gotas líquidas de una sustancia o de una mezcla en suspensión en un gas (en el aire por lo general)
- Vapor: forma gaseosa de una sustancia o de una mezcla liberada a partir de su estado líquido o sólido.
El polvo se forma generalmente por un proceso mecánico. Las nieblas se forman generalmente por condensación de vapores supersaturados o por el fraccionamiento físico de líquidos. El tamaño de polvos y nieblas oscila generalmente entre valores que van desde menos de UNO (1) hasta alrededor de CIEN MICRÓMETROS (100 |im);
f) Los valores para polvos y nieblas deberían revisarse para adaptarse a futuros cambios de las directrices de la Organización para la Cooperación y el Desarrollo Económico (OCDE) con respecto a las limitaciones técnicas en la generación, mantenimiento y medición de las concentraciones de polvos y nieblas en forma respirable;
g) Los criterios de la Categoría 5 se proponen identificar las sustancias que presenten un peligro relativamente bajo de toxicidad aguda, pero que en determinadas circunstancias puedan suponer un peligro para poblaciones vulnerables. La DL50 de esas sustancias se sitúa en el rango de DOS MIL A CINCO MIL MILIGRAMOS POR KILOGRAMO (2.000-5.000 mg/kg) de peso corporal y en dosis equivalentes para la inhalación. Los criterios específicos de la Categoría 5 son:
i. La sustancia se clasifica en esta categoría, si ya se dispone de información fidedigna que indique que la DL50 (CL50) corresponde al rango de valores de la Categoría 5 o cuando otros estudios con animales o sobre los efectos tóxicos agudos en seres humanos constituyen un motivo de preocupación para la salud humana;
ii. La sustancia se clasificará en esta categoría, mediante extrapolación, estimación o medición de datos, cuando no esté justificada su asignación a una categoría de mayor peligro; y:
- Se disponga de información fidedigna sobre la existencia de efectos tóxicos significativos en los seres humanos; o
- Se observe mortalidad en los ensayos sobre exposición por vía oral o cutánea o por inhalación, con valores hasta de la Categoría 4; o
- Cuando la opinión de los expertos confirme la aparición de síntomas clínicos de toxicidad significativos (excepto diarrea, piloerección o aspecto descuidado) en ensayos realizados con valores hasta de la Categoría 4; o
- Cuando tal opinión confirme la existencia de información fidedigna sobre efectos agudos potencialmente significativos procedentes de otros estudios con animales.
Tabla: Categorías y subcategorías de corrosión dermal.

Tabla: Elementos de corrosión/irritación dermal que deben figurar en una etiqueta.

Tabla: Categoría correspondiente a lesiones oculares graves/efectos irreversibles en los ojos.

Tabla: Categorías de efectos reversibles en los ojos.

Tabla: Elementos que deben figurar en la etiqueta para lesiones oculares graves/irritación ocular.

Tabla: Categoría y subcategorías de peligro para los sensibilizantes respiratorios.

Tabla: Categoría y subcategorías de peligro para los sensibilizantes dermales.

Tabla: Elementos que deben figurar en las etiquetas para sensibilizantes respiratorios y cutáneos.

Tabla: Categorías para las sustancias peligrosas para el medio ambiente acuático. Peligro a corto plazo (agudo).

Tabla: Elementos que deben figurar en las etiquetas de peligro para sustancias y mezclas peligrosas para el medio ambiente acuático.
PELIGRO A CORTO PLAZO (AGUDO) PARA EL MEDIO AMBIENTE ACUÁTICO

PELIGRO A LARGO PLAZO (CRÓNICO) PARA EL MEDIO AMBIENTE ACUÁTICO

Tabla: Categorías para las sustancias peligrosas para aves y elementos que deben figurar en la etiqueta.

Tabla: Categorías para las sustancias peligrosas para abejas y elementos que deben figurar en la etiqueta.

Tabla: Pictogramas de precaución para reducir riesgos al manipular, aplicar o almacenar un producto fitosanitario.
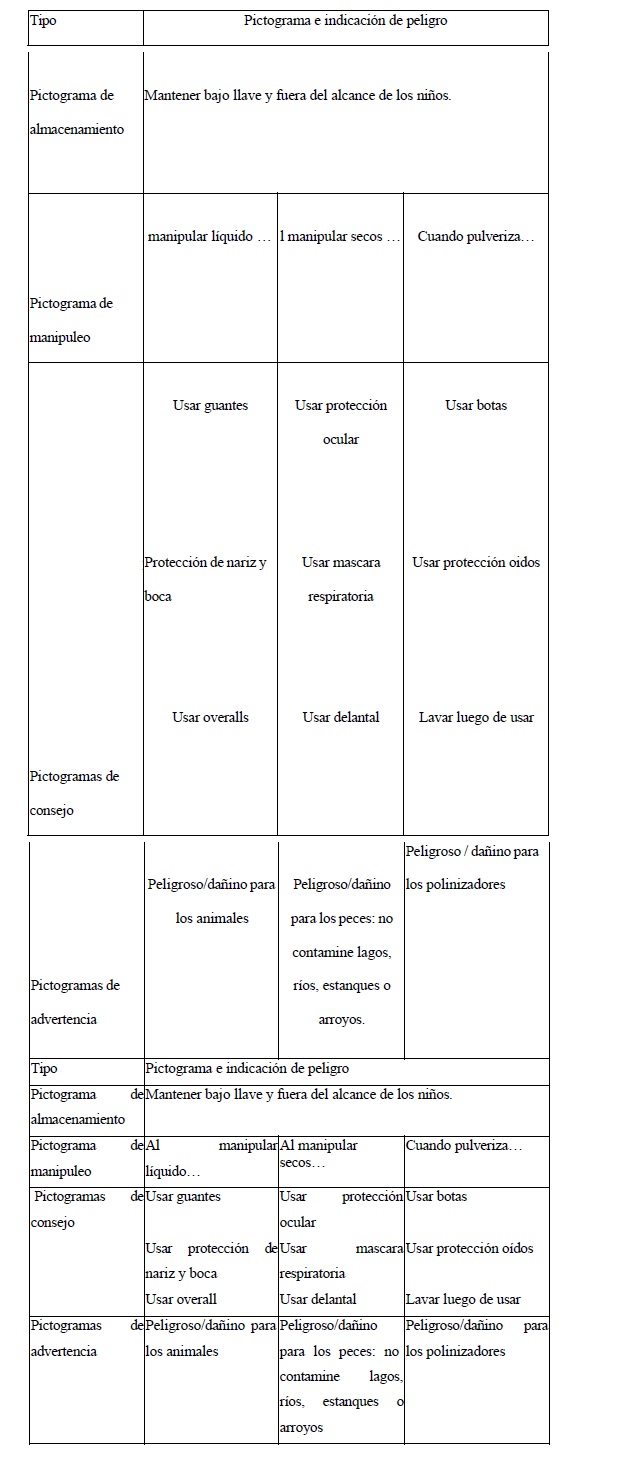
Tabla: Agrupación de Cultivos, mayores representativos y de cultivos menores para la REPÚBLICA ARGENTINA.

Tabla: Agrupación de Cultivos, mayores NO representativos y de cultivos menores para la REPÚBLICA ARGENTINA.

ANEXO I
(Anexo sustituido por art. 9° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
(Anexo sustituido por art. 9° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
AUTORIZACIÓN DE PERSONAS HUMANAS O JURÍDICAS Y ESTABLECIMIENTOS ELABORADORES
Todas las personas humanas o jurídicas que comercialicen, importen, para uso directo y/o los establecimientos que sinteticen o formulen productos fitosanitarios deberán solicitar una autorización con carácter de Declaración Jurada a través de la plataforma Trámites a Distancia (TAD), o la que en el futuro la reemplace.
Dicha autorización deberá contener los datos que a continuación se detallan:
Autorización de Persona Humana:
a) Nombre y apellido.
b) C.U.I.T. / C.U.I.L.
c) Teléfono.
d) Correo electrónico.
e) Nombre del Representante Legal y su correo electrónico.
Autorización de Personas Jurídicas:
a) Razón Social.
b) C.U.I.T. / C.U.I.L.
c) Dirección, teléfono y correo electrónico.
d) Nombre del Representante Legal y su correo electrónico.
Autorización de Establecimiento Elaborador:
a) Nombre y dirección de la compañía, persona humana o jurídica.
b) Nombre, dirección del establecimiento elaborador para el que se solicita y georreferenciación en coordenadas decimales (Decimal Degrees —DD—).
c) Para establecimientos situados en la República Argentina C.U.I.T. / C.U.I.L.
d) Teléfono y correo electrónico.
Cualquier modificación de las condiciones en las que fue otorgada la autorización, deberá ser comunicada a la Autoridad de Aplicación a través de sus sistemas informáticos.
La baja de la autorización podrá ser solicitada por la firma responsable del establecimiento o también producto de una sanción aplicada por el SENASA, como conclusión de un trámite sumarial del cual surge clara y fundadamente la gravedad del incumplimiento a la normativa vigente.
ANEXO II
(Anexo sustituido por art. 9° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
(Anexo sustituido por art. 9° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
AUTORIZACIÓN DE PREDIOS DE EXPERIMENTACIÓN DE PRODUCTOS FITOSANITARIOS EN ETAPAS TEMPRANAS DE DESARROLLO
REQUISITOS GENERALES
Las personas humanas o jurídicas interesadas en realizar experimentaciones a campo y/o en invernadero que impliquen la liberación al agroecosistema de productos fitosanitarios en etapas tempranas de desarrollo deberán solicitar una autorización con carácter de declaración jurada a través de la plataforma SIG-Trámites, o la que en el futuro la reemplace.
La autorización y las obligaciones del solicitante para la liberación de productos fitosanitarios en etapas tempranas comprende todas las etapas involucradas en el manejo bioseguro de los materiales en evaluación, desde el ingreso al país, el almacenamiento, el proceso de experimentación, la cosecha y la disposición final del material vegetal interviniente y los posibles remanentes de las sustancias evaluadas. Asimismo, comprende el monitoreo ambiental posterior del sitio de la liberación utilizado, por el período que se determine en la respectiva autorización.
Las personas humanas o jurídicas responsables del predio experimental tienen la obligación de facilitar el acceso a las instalaciones del predio y a los registros documentales, a los evaluadores del SENASA.
El SENASA otorgará dicha autorización en un plazo no mayor de TREINTA (30) días hábiles.
INFORMACIÓN ADMINISTRATIVA.
Solicitud de inscripción del predio de experimentación de productos fitosanitarios en etapas tempranas de desarrollo, con carácter de declaración jurada, firmada por el Propietario, Apoderado o Representante Legal y el Responsable Técnico con incumbencia profesional en la materia.
En dicha solicitud se deberá completar la siguiente información:
a) Nombre y Apellido/Razón Social.
b) C.U.I.T. / C.U.I.L.
c) Dirección del establecimiento y Georreferenciación.
d) Teléfono.
e) Correo electrónico.
f) Título profesional habilitante de los profesionales calificados involucrados directamente en actividades de investigación y experimentación con productos fitosanitarios.
CUERPO TÉCNICO
DESCRIPCIÓN DE LA ENTIDAD DE INVESTIGACIÓN:
Organigrama de la entidad, currículum vitae resumido y nota de responsabilidad técnica del Director de la unidad experimental, y de los profesionales calificados involucrados directamente en actividades de investigación y experimentación con productos fitosanitarios, desde la planificación hasta la emisión de informes técnicos.
DESCRIPCIÓN DEL ESTABLECIMIENTO:
- Croquis de la estación experimental con ubicación y memorial descriptivo que informe el área total y área de experimentación e investigación, estado de conservación de suelos, ubicación de cuerpos de agua y áreas aledañas.
-Croquis de acceso a la estación experimental, con las Coordenadas Geodésicas (Datum WGS84, expresados en grados y SEIS (6) decimales de grado), declarando las coordenadas geográficas del acceso al establecimiento y de los vértices que contengan la totalidad de la superficie del predio experimental.
-Croquis de ubicación de instalaciones/sectores: oficinas, laboratorios, invernáculos, zona de investigación/desarrollo de campo, depósitos. Las instalaciones deben contar con alambrado perimetral.
- Inventario de máquinas, equipos agrícolas, instalaciones físicas, medios técnicos y materiales. Las instalaciones deben contar con estación meteorológica, acceso a internet, telefonía celular y grupo electrógeno.
MANUAL DE PROCEDIMIENTOS:
-Flujo de las actividades a desarrollar, debiendo constar procedimientos de recibo, almacenamiento, transporte, manipuleo, utilización y disposición final de las muestras de sustancias experimentales.
- Detalle de la relación entre la zona geográfica elegida y el objetivo de la investigación.
- Indicación de los sistemas de registro del ingreso, flujo y destino de las muestras en proceso de experimentación temprana.
- Caracterización y tratamiento de los residuos especiales aprobados por la Autoridad Ambiental Competente a nivel municipal o provincial.
- Caracterización del tratamiento de envases vacíos y material descartable aprobado por la Autoridad Ambiental Competente a nivel municipal o provincial.
- Seguridad operativa en el manejo de sustancias químicas: debe indicar las políticas, actividades y normas internas de la empresa o institución en esta materia (equipos de seguridad, ropa protectora, control de efluentes, disposición final de muestras, destrucción de cultivos y otros).
- Plan de Control y Monitoreo Ambiental de los recursos suelo/agua/aire, y medidas de aislamiento y bioseguridad.
- Caracterización y tratamiento de los residuos de los materiales vegetales tratados.
- Plan de contingencias para el manejo de accidentes con sustancias químicas e incendios, elaborado por un profesional competente.
SISTEMA DE REGISTRO:
- Libro de estudio y de campo.
- Registro de ingreso, almacenamiento y disposición final de las muestras experimentales.
- Registro de aplicación, condiciones de aplicación y destrucción de material tratado.
- Registro de mantenimiento de equipos.
- Registro de capacitaciones del personal involucrado.
- Registro de almacenamiento y disposición final de los residuos generados en el predio experimental.
ARCHIVO:
- Archivo de plan de estudio, libros de campo y otros registros de la entidad.
ANEXO III
(Anexo sustituido por art. 9° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
(Anexo sustituido por art. 9° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
REGISTRO DE PRODUCTOS FITOSANITARIOS
Los productos fitosanitarios que se elaboren o formulen en la REPÚBLICA ARGENTINA o en otros países y no cuenten con el registro y la aprobación de uso agrícola por Autoridades Competentes del listado de países o grupos de países que se detallan en el Anexo IV deberán estar aprobados y registrados en el Registro Nacional de Productos Fitosanitarios, dependiente del SERVICIO NACIONAL DE SANIDAD Y CALIDAD AGROALIMENTARIA (SENASA) cumpliendo con lo establecido en el presente Anexo.
Los interesados que deseen registrar los productos deberán presentar un formulario de solicitud de registro dependiendo del tipo de producto a registrar, a través de la plataforma SIG-Trámites o la que en el futuro la reemplace, y la documentación correspondiente según el tipo de producto.
El SENASA procederá a evaluar la información presentada y expedirse en un plazo no mayor a CIENTO OCHENTA (180) días hábiles.
A los efectos de la presente norma los productos a registrar estarán encuadrados en DOS (2) categorías dependiendo del tipo de producto a registrar:
SUSTANCIAS ACTIVAS: se entiende por sustancia activa al componente principal de un producto fitosanitario que tiene la capacidad de ejercer un efecto biológico. Esta puede ser grado técnico nueva o equivalente.
SUSTANCIAS ACTIVAS NUEVAS: se considera sustancia activa grado técnico nueva a la que no ha sido registrada en el país.
En los casos en que se desee registrar este tipo de sustancias se deberá proceder al registro completo detallado en los apartados I a VI.
SUSTANCIAS ACTIVAS EQUIVALENTES: se considera sustancia activa equivalente cuando su equivalencia ha sido demostrada respecto de otras ya registradas en el país.
Las sustancias activas de diferentes fabricantes o de diferentes procesos de síntesis del mismo fabricante son equivalentes si:
- La sustancia activa tiene una concentración declarada mayor o igual a la de la referencia.
- El máximo nivel de impurezas no relevantes no se incrementa por más del CINCUENTA POR CIENTO (50 %) (relativo al máximo nivel en la referencia), o el nivel absoluto no se incrementa en más del CERO COMA TRES POR CIENTO PESO EN PESO (0,3 %p/p), considerando el que represente el mayor nivel de incremento.
- No se presentan nuevas impurezas mayores a CERO COMA UNO POR CIENTO PESO EN PESO (0,1 %p/p).
- No se encuentran impurezas relevantes por encima de los límites establecidos en la Resolución N° 481 del 27 de octubre de 2014 del citado Servicio Nacional y sus modificatorias, o en las especificaciones de la Organización de las Naciones Unidas para la Alimentación y la Agricultura (FAO).
En los casos en que se desee registrar este tipo de sustancias activas equivalentes, se deberán cumplimentar los apartados detallados en los puntos I a III.
PRODUCTO FORMULADO: se entiende por producto formulado a la mezcla de sustancias activas y otras sustancias, que cumple la función de proteger a las plantas contra plagas, enfermedades y malezas.
Los coadyuvantes incluidos como componentes de una formulación no serán pasibles de registro, pero deberán usarse para la elaboración o fabricación de productos fitosanitarios aquellos que no se encuentren alcanzados en la Resolución N° RESOL-2019-32-APN-PRES#SENASA del 17 de enero de 2019 del mencionado Servicio Nacional, o la que en un futuro la reemplace.
A fin de realizar la inscripción de fitosanitarios al registro, los requerimientos para las diferentes categorías son los siguientes:
A - SUSTANCIAS ACTIVAS
I - INFORMACIÓN ADMINISTRATIVA
Formulario de solicitud de registro, dependiendo del tipo de producto a registrar, firmado por el Apoderado o Representante Legal y el Responsable Técnico con incumbencia profesional en la materia.
Patrones: en caso de que el SENASA así lo requiera para control, la empresa registrante deberá aportar los patrones analíticos de la sustancia activa e impurezas, en las condiciones que establezca la Dirección General de Laboratorios y Control Técnico (DGLCyT) del SENASA.
Hoja de Datos de Seguridad: de acuerdo con la versión del Manual de las Naciones Unidas del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA), adoptada en la presente norma.
Informes técnicos de análisis de riesgo toxicológico y ecotoxicológico de nuevos principios activos de uso agrícola, firmados por los equipos interdisciplinarios de profesionales independientes especializados en toxicología y ecotoxicología del SENASA.
Si se desea registrar una sustancia activa grado técnico equivalente que se produzca en el país o que se importe desde países no incluidos en el Anexo IV, para acreditar dicha equivalencia se deberá presentar ante el SENASA el formulario de solicitud de registro, junto con toda la documentación técnica y administrativa requerida.
II - GENERALIDADES
Protocolos y estudios de Productos Fitosanitarios
Los estudios biológicos para la producción de datos toxicológicos y ecotoxicológicos y los laboratorios que realicen propiedades físicas y químicas, y determinaciones analíticas de residuos de principios activos en matrices vegetales y ambientales con fines de registro, revalidación, revaluación o monitoreo de productos fitosanitarios, deberán incluir la documentación respaldatoria correspondiente al monitoreo de Buenas Prácticas de Laboratorio (BPL), desarrolladas por la Organización para la Cooperación y el Desarrollo Económico (OCDE), emitida por un Organismo de Acreditación de reconocimiento internacional. Asimismo, los ensayos de residuos de principios activos deberán encuadrarse en la Directiva OCDE sobre aplicación de las BPL para los estudios de campo.
Los protocolos de los estudios biológicos para la producción de datos toxicológicos y de propiedades físicas y químicas y las determinaciones analíticas de residuos de principios activos deben realizarse mediante los protocolos correspondientes a los organismos y cuerpos normativos que protocolizan ensayos y procedimientos de laboratorio para la obtención de datos con fines de registro, a saber:
APVMA: AUSTRALIAN PESTICIDES AND VETERINARY MEDICINES AUTHORITY
• CIPAC: COLLABORATIVE INTERNATIONAL PESTICIDES ANALYTICAL COUNCIL
• EEC/EU: EUROPEAN ECONOMIC COMMUNITY/EUROPEAN UNION
• EFSA: EUROPEAN FOOD SAFETY AUTHORITY
• EPA: UNITED STATES ENVIRONMENTAL PROTECTION AGENCY
• EURACHEM: EUROPEAN COLLABORATION ON CHEMICAL MEASUREMENT
• FDA: UNITED STATES FOOD AND DRUG ADMINISTRATION
• FFDCA: FEDERAL FOOD DRUG AND COSMETIC ACT
• FIFRA: FEDERAL INSECTICIDE, FUNGICIDE AND RODENTICIDE ACT
• ISO: INTERNATIONAL STANDARD ORGANIZATION
• IUPAC: INTERNATIONAL UNION OF PURE AND APPLIED CHEMISTRY
• OECD: ORGANIZATION FOR ECONOMIC COOPERATION AND DEVELOPMENT
• OMS: WORLD HEALTH ORGANIZATION
• OPPTS: OFFICE OF PREVENTION, PESTICIDES AND TOXIC SUBSTANCES
Otros protocolos provenientes de organismos o cuerpos normativos diferentes a los que figuran en el presente Manual deberán ser consultados previamente al SENASA.
Clasificación toxicológica
Se adoptará como clasificación toxicológica la del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA/GHS), utilizando para tal fin la información sobre toxicología en mamíferos y no mamíferos de la sustancia activa o del producto formulado, según corresponda.
A los fines de comunicar el peligro en los marbetes de productos fitosanitarios y las hojas de datos de seguridad de estos, se deberá proceder de acuerdo con los criterios definidos en la Versión ST/SG/AC.10/30/Rev.9 del Manual de las Naciones Unidas del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA), y en función de la normativa de etiquetado de productos fitosanitarios vigente en la REPÚBLICA ARGENTINA, versión revisable al efecto del presente manual cada CINCO (5) años.
Accesibilidad de la información de registro
La información confidencial y los datos de pruebas serán provistos por el registrante, distinguiendo claramente entre qué es información confidencial y qué son datos de pruebas, considerando: Información confidencial: la información correspondiente a la identidad, composición, proceso de síntesis, proceso de formulación y otros secretos industriales y comerciales.
Datos de pruebas: datos de ensayos toxicológicos, ecotoxicológicos, de residuos y propiedades físicas y químicas.
Respecto a la información confidencial o a los datos no divulgados requeridos para su evaluación, el SENASA tomará las medidas necesarias para su salvaguarda contra todo uso comercial deshonesto y evitar que dicha información sea revelada o transferida. A tales efectos, el SENASA se encargará de proveer las instalaciones y los medios necesarios para garantizar la seguridad y una adecuada gestión de la información.
A tal fin, la información confidencial y los datos no publicados recibidos serán resguardados informáticamente en los sistemas que disponga especialmente el SENASA, y será proporcionada a los técnicos evaluadores a tal fin, no pudiendo estos efectuar copias ni totales ni parciales, ni compartir dicha información o darla a conocer a terceros.
Los técnicos evaluadores (funcionarios internos o auxiliares) dejarán constancia escrita de que aceptan las condiciones de confidencialidad en que deberán manejar el material informativo que se les provea, bajo declaración jurada.
Concluida la evaluación, la información confidencial y los datos no publicados serán resguardados siguiendo las medidas de seguridad dispuestas. El SENASA podrá disponer su entrega al administrado para que éste los tenga en guarda. En este último caso, la información confidencial y los datos no publicados deben estar disponibles en el caso que el SENASA lo solicite.
La información confidencial de las sustancias activas grado técnico consideradas de referencia para la determinación de equivalencia quedan en guarda del SENASA y no están alcanzadas por lo dispuesto en el párrafo previo respecto de su entrega al administrado.
Cuando los datos de ensayos y pruebas sobre seguridad y eficacia, resguardados en el Organismo, hubieran caído en el dominio público en cualquier país por la publicación de cualquiera de los datos protegidos, la presentación de todos o parte de estos datos en medios científicos o académicos o por cualquier otro medio de publicación, entonces estos dejarán de ser archivados mediante las medidas de seguridad dispuestas por el SENASA.
Los expedientes técnicos de registro son reservados de acuerdo con el marco previsto en el Artículo 38 del Decreto N° 1.759 del 3 de abril de 1972, sus modificatorios y complementarios. Su vista queda reservada al personal y los auxiliares de este Servicio Nacional afectados al procedimiento de registro, y a las personas fehacientemente autorizadas por el administrado titular del registro.
El SENASA utilizará la información suministrada del modo antes descrito a los efectos de los registros de productos fitosanitarios, observando el marco legal que impone la Ley N° 24.766 y normas complementarias.
Quedan expresamente exceptuados de la confidencialidad:
a) Nombre, contenido y origen de principios activos en productos formulados, establecimientos elaboradores y empresas registrantes.
b) Métodos y recomendaciones de transporte, almacenaje, tratamientos de incendio y otros riesgos.
c) Medios de disposición de envases.
d) Procedimientos de descontaminación.
e) Primeros auxilios y ayuda médica en caso de daño a las personas.
f) Método de análisis de residuos.
g) Método de análisis de las impurezas de importancia toxicológica o ecotoxicológica (denominadas de declaración obligatoria).
h) La información contenida en la Hoja de Datos de Seguridad.
i) Toda información que haya caído en el dominio público.
Quienes requieran la información referida en los incisos f) y g), deberán hacerlo mediante nota, expresando el motivo del requerimiento, la que será registrada y archivada.
El personal afectado a los procedimientos de registro de productos fitosanitarios se encuentra comprendido en los mandatos de los Artículos 3°, 12 y 13 de la Ley N° 24.766, por lo que deberá abstenerse de usar y de revelar sin causa justificada o sin consentimiento del registrante la información en cuestión, bajo apercibimiento de las sanciones que la misma norma prevé.
III - INFORMACIÓN CONFIDENCIAL (tanto para sustancias activas nuevas, como equivalentes)
a.- Certificado de Origen, el cual debe ser original y emitido por el establecimiento elaborador.
Debe incluir:
Identificación de la sustancia activa.
Contenido mínimo declarado porcentaje PESO EN PESO (p/p).
Nombre y localización del establecimiento elaborador.
Clave o número con el que la Autoridad Competente del país de origen identifica al establecimiento.
Nombre y dirección de la empresa que registrará el producto ante SENASA. En caso de que no figure esta información se deberá remitir una nota emitida por el establecimiento elaborador donde conste la autorización para realizar el registro del producto en el país.
b - Declaración de composición cuali-cuantitativa firmada por el establecimiento elaborador o por la empresa registrante con carácter de declaración jurada.
Debe incluir:
Nombre químico según criterios internacionales [Unión Internacional de Química Pura y Aplicada (IUPAC) o Chemical Abstract Service (CAS)] y número CAS (cuando esté disponible) de la sustancia activa y sus impurezas asociadas.
Concentración mínima de la sustancia activa.
Concentración máxima de cada impureza mayor o igual a CERO COMA UNO POR CIENTO PESO EN PESO (0,1 %p/p).
Concentración máxima de impurezas relevantes: el límite establecido por la normativa vigente y de no existir, el límite de detección del método utilizado.
La fracción no identificada de la sustancia activa no podrá superar el DOS POR CIENTO (2 %) por lote y deberá componerse solo de impurezas con concentración inferior al CERO COMA UNO POR CIENTO PESO EN PESO (0,1 %p/p), excluyendo impurezas relevantes.
La concentración declarada se basará en el análisis de muestras representativas de al menos CINCO (5) lotes de síntesis, asegurando que en todos los casos la concentración obtenida sea igual o superior al límite inferior establecido.
La concentración mínima de sustancia activa se determinará mediante un análisis estadístico, por ejemplo, la media menos TRES (3) desvíos estándar. Si el valor declarado es inferior, deberá justificarse el criterio estadístico o la razón para su reducción.
La concentración máxima de impurezas se obtendrá con un análisis similar, tomando la media más TRES (3) desvíos estándar. Si el valor declarado es superior, deberá explicarse el criterio utilizado o la justificación del aumento.
c - Estudio y cuantificación en CINCO (5) lotes
Debe presentarse un estudio completo de al menos CINCO (5) lotes de síntesis independientes, realizado por un laboratorio conforme a las Buenas Prácticas de Laboratorio (BPL), debidamente firmado y fechado. Si los sponsors del estudio no coinciden con la empresa registrante, deberá incluirse una autorización para el uso de la información.
El reporte debe contener:
Índice de contenidos.
Nombre y localización del establecimiento elaborador.
Cuadro resumen de resultados.
Métodos de análisis con todas las ecuaciones necesarias para reproducir los cálculos.
Resultados, incluyendo cromatogramas representativos de cada lote, del blanco y de los patrones analíticos.
En caso de usar curvas de calibración, los gráficos con títulos, ejes (con magnitudes y unidades) y ecuación de la recta.
Datos en formato de cuadro para reproducir cálculos, dentro del informe o como anexo. Cromatogramas con encabezado (fecha, hora, descripción) y reporte (áreas y tiempos de retención). Certificados de análisis de los patrones utilizados.
Certificados de análisis de los lotes analizados, emitidos y firmados por el fabricante, incluyendo nombre y dirección del establecimiento elaborador, fecha de elaboración y vencimiento. Si la dirección no figura en el certificado, deberá presentarse una declaración jurada con dicha información.
Certificado BPL del Laboratorio.
Además, el análisis de impurezas debe realizarse en los mismos lotes de síntesis, incorporando:
Un screening con detector universal para visualizar impurezas en concentraciones menores a CERO COMA UNO POR CIENTO PESO EN PESO (0,1 % p/p), ajustado a la impureza significativa de menor concentración.
Certificados de estándares con reporte de cuantificación e identificación. En caso de patrones secundarios, cuantificación contra el patrón primario y su certificado de análisis. Si se utilizan patrones de sustancias análogas, deberá presentarse una justificación técnica demostrando la analogía molecular.
d - Análisis de identidad de la sustancia activa e impurezas
La identidad de la sustancia activa deberá confirmarse mediante determinaciones analíticas que permitan establecer fehacientemente su estructura química y, si corresponde, su configuración molecular. Para uno de los CINCO (5) lotes analizados, se presentarán al menos DOS (2) espectros entre: Espectroscopía Infrarroja (IR), Resonancia Magnética Nuclear (RMN) y Espectrometría de Masas (MS). Se incluirá una discusión clara y concisa sobre la interpretación de los espectros, demostrando la identidad de la sustancia activa en grado técnico o comparándolos con los espectros del estándar analítico.
Asimismo, la identidad de todas las impurezas, o de grupos de impurezas relacionadas, deberá ser demostrada mediante análisis espectrométricos, espectroscópicos y/o químicos que permitan identificarlas inequívocamente. Cada impureza deberá estar identificada en al menos UNO (1) de los CINCO (5) lotes analizados, presentando un espectro de masa o, en caso de contar con patrones, una comparación de espectros espectroscopia ultravioleta-visible (UV-vis) y tiempos de retención. Se acompañará la información con explicaciones claras y concisas que justifiquen la identificación de cada impureza o grupo de impurezas relacionadas.
e - Métodos de análisis
El registrante debe proveer los métodos analíticos utilizados para la determinación, tanto de la sustancia activa como de las impurezas. Deberán incluir la descripción completa y los parámetros de validación respectivos (especificidad, linealidad, recuperación, precisión, límites de detección y de cuantificación en el caso de las impurezas). Además de lo especificado en los contenidos mínimos establecidos en el ítem 3, el reporte debe incluir cromatogramas representativos correspondientes a cada uno de los parámetros estudiados.
f- Proceso de síntesis
Debe proveerse la siguiente información emitida y firmada por el fabricante o la empresa registrante:
Nombre y localización del establecimiento elaborador que interviene en el proceso.
Caracterización general del proceso: deberá consistir en una descripción completa y detallada de cada una de las etapas que constituyen el proceso, incluyendo en forma explícita las reacciones químicas que tienen lugar en cada una de ellas y sus condiciones de presión y temperatura. Cuando corresponda, incluir otros parámetros controlados [por ejemplo, potencial de hidrógeno (pH), humedad, etcétera]. Detallar los criterios usados para dar por finalizada cada etapa de reacción.
Diagrama de flujo.
Identificación de todos los reactivos usados en el proceso, con las especificaciones requeridas.
Descripción de los equipos usados y sus características.
g.- Justificación de la presencia de impurezas
El fabricante deberá suministrar la explicación pertinente sobre el origen y la formación de las impurezas que pueden estar presentes en el producto final, especificando la estructura molecular y el nombre químico para cada una de ellas. Debe indicar las reacciones químicas que describen su formación, las que deben estar sustentadas en mecanismos reconocidos y probados, y compatibles con las reacciones que constituyen el proceso de síntesis.
Si el SENASA considera que una impureza declarada, o que sea probable que esté presente en el grado técnico puede ser considerada relevante, deberá solicitar a la empresa registrante que remita la discusión que fundamente su condición así como la concentración en que puede encontrarse.
En caso de que el sistema de vigilancia post registro de activos demuestre inconsistencias entre la información declarada y los análisis efectuados por la Dirección de Agroquímicos y Biológicos (DAyB) en las muestras oficiales, el SENASA se reserva el derecho de solicitar documentación adicional o dar de baja el registro.
h Sobre los efectos tóxicos en especies mamíferas (solo para sustancias activas equivalentes)
Si la evaluación de equivalencia química determina la presencia de nuevas impurezas relevantes o impurezas excedidas respecto a la referencia, el registrante deberá justificar dichas impurezas conforme a los procedimientos establecidos y presentar un test de mutagénesis junto con un informe toxicológico. Este deberá demostrar que los efectos subcrónicos y/o crónicos no son significativamente superiores a los de la sustancia parental, pudiendo basarse en estudios de relaciones cuantitativas estructura-actividad (QSAR) en DOS (2) parámetros toxicológicos crónicos relevantes.
Para evaluar los posibles efectos dañinos que deben consignarse en la etiqueta elemental del grado técnico y en la Hoja de Datos de Seguridad, se tomará como referencia la información de la sustancia parental una vez establecida la equivalencia, en concordancia con la versión adoptada por la Autoridad Competente del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA) de las Naciones Unidas.
IV - CUERPO TÉCNICO
SOBRE LAS PROPIEDADES FÍSICAS Y QUÍMICAS:
Aspecto
Estado físico
Color
Densidad
Presión de vapor
Solubilidad en agua
Solubilidad en disolventes orgánicos
Punto de inflamación
Propiedades oxidantes
Potencial de hidrógeno (pH), si la sustancia activa grado técnico es soluble o dispersable en agua.
Para el caso de sustancias activas grado técnico equivalentes, el cuerpo técnico debe incluir también la siguiente información:
SOBRE LA IDENTIDAD:
Fabricante.
Nombre común: aceptado por la Organización Internacional de Estandarización (ISO), o equivalente (si está disponible).
Nombre químico: aceptado o propuesto por IUPAC o CAS (si está disponible).
Número CAS, CIPAC y/o número de código experimental.
Fórmula empírica, peso molecular.
Fórmula estructural.
Grupo químico.
Concentración de sustancia activa.
SOBRE LOS ASPECTOS RELACIONADOS A SU USO.
1. Organismos nocivos controlados.
2. Modo de acción sobre los organismos nocivos y sobre las plantas.
3. Ámbito de aplicación previsto.
4. Condiciones fitosanitarias y ambientales para ser usado.
SOBRE LOS MÉTODOS ANALÍTICOS (para la determinación de residuos en cultivos tratados, subproductos de origen vegetal, alimentos procesados, suelo y agua)
Se incluirá la tasa de recuperación y los límites de sensibilidad metodológica.
Estos métodos serán requeridos si los usos propuestos para el producto requieren el establecimiento de una tolerancia.
Métodos analíticos para la determinación en el aire: Estos serán requeridos para productos volátiles. Métodos analíticos para la determinación en tejidos y fluidos animales o humanos: Estos métodos serán requeridos cuando las plantas tratadas se destinen a la alimentación animal.
V- RESIDUOS EN PRODUCTOS TRATADOS (ÚNICAMENTE REQUERIDO PARA SUSTANCIAS ACTIVAS DE GRADO TÉCNICO NUEVAS y AMPLIACIONES DE NUEVO USO)
Los estudios serán requeridos si los usos propuestos exigen el establecimiento de una tolerancia. El cálculo del Límite Máximo de Residuos (LMR) se realizará utilizando la calculadora de la OCDE, basada en los datos disponibles.
a. Metabolismo en Vegetales (para sustancias activas nuevas)
Debe identificarse la degradación y reacción de metabolitos en plantas o productos tratados. Se requiere un estudio de metabolismo en un cultivo representativo de cada categoría o grupo de cultivos para los que se propone el uso, conforme a las Directrices de la OCDE u otras reconocidas. Para extrapolar el metabolismo a todos los grupos, se realizarán estudios en al menos TRES (3) de las CINCO (5) categorías de cultivos; si los resultados son comparables, no serán necesarios estudios adicionales.
b. Comportamiento de Residuos (para sustancias activas nuevas y ampliaciones de uso en nuevos cultivos, incremento de dosis, reducción del tiempo de carencia)
Para usos que requieran la fijación de tolerancias, se realizarán ensayos de residuos en cultivos para establecer un LMR. Estos estudios deberán cumplir con Buenas Prácticas de Laboratorio (BPL) en sus fases analíticas y de campo. En caso de requerirse estudios locales, la fase de campo deberá realizarse en la REPÚBLICA ARGENTINA con certificación BPL.
Para aquellos usos que requieran la fijación de tolerancias, se requerirán ensayos de residuos en cultivos a fin de establecer un Límite Máximo de Residuos para el cultivo o grupo de cultivos para el que se requiera autorizar el uso de la sustancia activa.
Los estudios de residuos de productos fitosanitarios deben ser realizados por entidades que hayan certificado el cumplimiento de Buenas Prácticas de Laboratorio (BPL), tanto en sus fases analíticas, como de campo.
Para aquellos casos en los que se requiera un número mínimo de estudios de residuos locales, la fase de campo deberá realizarse en el territorio Argentino por entidades que hayan certificado el cumplimiento de BPL.
El plan de ensayos deberá ajustarse a este Manual y los ensayos de campo en Argentina deberán emplear variedades representativas en la región evaluada. Para ensayos realizados en la misma campaña, los lotes experimentales deben distanciarse al menos VEINTE (20) kilómetros y tener un desfase de QUINCE (15) días.
Los ensayos deben respetar la práctica agrícola crítica indicada en la etiqueta del producto formulado: dosis máxima, número de aplicaciones, intervalo entre aplicaciones y período de carencia. Previa justificación técnica, se aceptará una variación en más o en menos de un VEINTICINCO POR CIENTO (±25 %) en un parámetro de la práctica agrícola crítica y el criterio de proporcionalidad de la dosis máxima, siguiendo directrices de la OCDE.
Cuando se requieran curvas de degradación, estas deberán incluir al menos TRES (3) puntos y abarcar el período de carencia. Si el residuo es menor al Límite de Cuantificación (LOQ), no será necesario realizar curvas de degradación.
Los ensayos destinados a establecer un periodo de seguridad (usos en postcosecha), no requerirán curvas de disipación, pero las muestras deben tomarse dentro del período de seguridad propuesto. El producto formulado utilizado en el ensayo debe caracterizarse bajo principios BPL, con Certificado de Análisis incluido en el informe. Las muestras deben analizarse en un máximo de TREINTA (30) días tras la recolección; en caso contrario, deberá presentarse un estudio de estabilidad de la molécula y sus metabolitos en una matriz representativa.
Productos exentos de fijación de LMR:
• Tratamiento de semillas y órganos de propagación no destinados al consumo.
• Aplicaciones en cultivos florales, ornamentales o áreas no cultivadas.
• Arbusticidas de acción tópica, preservadores de madera y hormiguicidas no aplicables al vegetal.
• Feromonas, atractivos, repelentes, rodenticidas y coadyuvantes.
• Biocontroladores
• Otros productos definidos con base técnica y científica.
c. Residuos en Cultivos Rotacionales (Nuevas sustancias activas - Condicionalmente Requeridos)
Si la sustancia activa se usa en cultivos anuales y sus metabolitos persisten en el suelo, pueden exigirse estudios de metabolismo en cultivos de rotación.
Si estos estudios indican residuos de la sustancia activa o metabolitos MAYOR A CERO COMA CERO UN MILIGRAMOS POR KILOGRAMO (> 0,01 mg/kg) en alimentos o MAYOR O IGUAL A CERO COMA CERO CINCO MILIGRAMOS POR KILOGRAMO (> 0,05 mg/kg) en forrajes, se seguirán directrices de la OCDE. Se aceptarán estudios realizados en otros países.
VI - EFECTOS TÓXICOS EN ESPECIES MAMÍFERAS. (ÚNICAMENTE REQUERIDO PARA SUSTANCIAS ACTIVAS DE GRADO TÉCNICO NUEVAS)
Se deberá aplicar un enfoque basado en toda la información disponible (peso de la evidencia) para determinar si un estudio estándar, un estudio con parámetros adicionales o un método alternativo al uso de animales, permite evaluar adecuadamente el peligro para la salud humana o justificar una exención de estudios específicos.
Los estudios deberán seguir las Test Guidelines y Guidance Documents de la OCDE u otros reconocidos internacionalmente.
a - TOXICIDAD AGUDA
Se requerirá el estudio de toxicidad oral aguda (DL50 oral) en todos los casos, excepto cuando la sustancia activa de grado técnico sea un gas o altamente volátil.
El estudio de toxicidad cutánea aguda (DL50 cutánea) será obligatorio al momento de la inscripción definitiva, salvo que el producto formulado sea un gas, altamente volátil o corrosivo para la piel, o presente un pH inferior a DOS (2) o superior a ONCE COMA CINCO (11,5).
El estudio de toxicidad aguda por inhalación (LC50 inhalación) será requerido cuando la sustancia activa de grado técnico tenga una presión de vapor superior a 10-2 Pa a 20 °C, sea un polvo con una proporción significativa de partículas con diámetro menor a 50 p,m [más del UNO POR CIENTO (1 %) en peso] o se incluya en productos en polvo o que se apliquen por pulverización.
El estudio de irritación cutánea in vivo no deberá realizarse cuando la sustancia activa de grado técnico sea un gas o altamente volátil, sea corrosiva para la piel, presente un pH inferior a DOS (2) o superior a ONCE COMA CINCO (11,5), cuente con información suficiente que permita clasificarla como corrosiva para la piel o irritante ocular, o haya sido clasificada como muy tóxica por vía dérmica.
El estudio de irritación ocular será obligatorio, salvo que la sustancia activa de grado técnico sea corrosiva o severamente irritante para la piel, presente un pH inferior a DOS (2) o superior a ONCE COMA CINCO (11,5) o haya sido clasificada como muy tóxica por vía dérmica.
El estudio de sensibilización cutánea será requerido, excepto cuando la sustancia activa sea un sensibilizante conocido.
En cuanto a la mutagenicidad, será obligatorio realizar un estudio in vitro en el momento del registro experimental. Si la empresa registrante no dispone de estudios toxicológicos in vivo, podrá presentar estudios alternativos in vitro, in silico u otros métodos reconocidos por autoridades regulatorias internacionales con convergencia normativa entre la REPÚBLICA ARGENTINA, para el registro de productos fitosanitarios. En estos casos, deberá solicitarse su aceptación mediante una nota con una justificación técnico-científica del uso de dichos estudios.
b - TOXICIDAD SUBCRÓNICA (corto plazo/medio plazo)
- Cuando esté disponible, se deberán comunicar los estudios de toxicidad oral acumulativa con una duración de VEINTIOCHO (28) días.
- El estudio de toxicidad oral a corto plazo en roedores deberá realizarse con una duración de NOVENTA (90) días, utilizando ratas como especie de prueba, salvo que se justifique el uso de otra especie. Para no roedores, se deberá presentar un estudio de toxicidad de NOVENTA (90) días en perros. En ambos casos, deberán evaluarse el potencial neurotóxico, los efectos inmunotóxicos, la genotoxicidad a través de la formación de micronúcleos y los posibles efectos sobre el sistema hormonal.
- El estudio de toxicidad inhalatoria con exposición repetida durante VEINTIOCHO (28) o NOVENTA (90) días será requerido condicionalmente para sustancias activas volátiles con una presión de vapor superior a 10“2 Pascales, cuando sea necesario para refinar la evaluación del riesgo ocupacional.
El estudio de toxicidad dérmica con administración repetida durante VEINTIÚN (21), VEINTIOCHO (28) o NOVENTA (90) días será requerido condicionalmente para el refinamiento de la evaluación del riesgo ocupacional, en caso de que los estudios de toxicología aguda dérmica o los valores por defecto de penetración dérmica no sean suficientes para su determinación.
c - TOXICIDAD CRÓNICA A LARGO PLAZO Y CARCINOGENICIDAD.
Se requerirá un estudio de toxicidad oral a largo plazo y un estudio de carcinogenicidad a largo plazo de la sustancia activa, utilizando ratas como especie de prueba; cuando sea posible, estos estudios se combinarán.
Las duraciones mínimas de estudio aceptables son:
Estudio de carcinogenicidad en ratones: 18 meses.
Estudio combinado toxicidad oral a largo plazo y carcinogenicidad en ratas: 12 - 24 meses.
Se requerirá un segundo estudio de carcinogenicidad de la sustancia activa utilizando ratón como especie de ensayo, a menos que pueda justificarse científicamente que no es necesario. En tales casos, se pueden utilizar modelos alternativos de carcinogenicidad validados científicamente.
Se deben remitir los datos experimentales, incluyendo la determinación del posible mecanismo de carcinogenicidad implicado y su relevancia para los seres humanos, cuando se considere que el modo de acción de la carcinogenicidad no es genotóxico.
d.- MUTAGENICIDAD
Los estudios de mutagenicidad in vitro deberán realizarse según corresponda, incluyendo al menos uno de los siguientes ensayos: ensayo bacteriano para mutación genética, prueba combinada para aberraciones cromosómicas estructurales y numéricas en células de mamífero, o prueba de mutación genética en células de mamíferos.
Los estudios de mutagenicidad in vivo en células somáticas solo serán requeridos si al menos uno de los estudios in vitro arroja un resultado positivo. En tal caso, deberá realizarse al menos un estudio in vivo que evalúe específicamente el efecto de genotoxicidad identificado en los ensayos in vitro. Si se requiere un estudio de micronúcleos in vivo, este podrá integrarse dentro de un estudio de dosis repetida.
La necesidad de estudios in vivo en células germinales se evaluará caso por caso, en función de los resultados obtenidos en células somáticas, considerando la toxicocinética de la sustancia, su uso y la exposición anticipada.
e - EFECTOS SOBRE LA REPRODUCCIÓN
Para evaluar los efectos sobre la reproducción, se recomienda la realización de un estudio combinado de toxicidad reproductiva en ratas, utilizando un estudio de dos generaciones o de una generación extendida como protocolo base. Este estudio deberá incluir criterios adicionales de valoración y evaluaciones funcionales en animales inmaduros.
Los estudios de toxicidad del desarrollo serán obligatorios y deberán realizarse en ratas y conejos por vía oral.
f - METABOLISMO EN MAMÍFEROS
Se deberán realizar estudios in vitro comparativos del metabolismo de la sustancia activa entre especies animales y humanos.
Los estudios in vivo en mamíferos deberán proporcionar información sobre la cinética de la sustancia activa y sus metabolitos en especies relevantes, considerando:
Una dosis oral única en niveles de dosis baja y alta.
Una dosis intravenosa o, si está disponible, una dosis oral única con evaluación de la excreción biliar en nivel de dosis baja.
Una dosis repetida.
Para el refinamiento de la evaluación de riesgo ocupacional, podrá requerirse un estudio de penetración dérmica in vitro en piel humana a fin de determinar la biodisponibilidad por vía dérmica.
Asimismo, deberá incluirse una descripción detallada de las rutas metabólicas de la sustancia activa y sus metabolitos.
g - ESTUDIOS DE NEUROTOXICIDAD EN ROEDORES
Los estudios serán condicionalmente requeridos a partir de un enfoque basado en el peso de la evidencia, considerando que:
La sustancia causa efectos neurológicos en animales adultos (es decir, signos clínicos de neurotoxicidad, neuropatología, alteraciones funcionales o de comportamiento).
La sustancia causa efectos neurológicos en animales en desarrollo, después de la exposición pre y posnatal (es decir, malformaciones del sistema nervioso o neuropatía, cambios de peso cerebral en la descendencia, alteraciones funcionales o de comportamiento en la descendencia). la sustancia evoque un mecanismo que está asociado con efectos adversos sobre el desarrollo del sistema nervioso, en comparación con neurotóxicos conocidos, respuestas de neurorreceptores o neurotransmisores alterados.
Se recomienda el uso de un enfoque combinado que utilice el estudio de reproducción de DOS (2) generaciones en roedores o de UNA (1) generación extendida, como protocolo básico para la evaluación de efectos de neurotoxicidad y evaluaciones funcionales en animales inmaduros.
h- OTROS ESTUDIOS TOXICOLÓGICOS
Cuando los metabolitos de la sustancia activa generados en plantas, productos animales, suelo o agua subterránea difieran de aquellos identificados en los estudios toxicológicos en animales, o se detecten en bajas proporciones en estos últimos, se requerirán estudios adicionales. La necesidad de estos estudios se evaluará caso por caso, considerando la cantidad y la estructura química del metabolito en relación con la sustancia parental.
En lo que respecta a la Información Médica Obligatoria, se deberá presentar información sobre: Diagnóstico y síntomas de intoxicación.
Observaciones sobre sensibilización y alergización.
Efectos tóxicos de los metabolitos provenientes de vegetales tratados.
Información médica complementaria, cuando esté disponible.
Elaboración de una ficha médica definitiva.
i - INFORMACIÓN CON RESPECTO A LA SEGURIDAD
Se deberán establecer procedimientos específicos para la destrucción de la sustancia activa y su descontaminación, así como la evaluación de posibles métodos de recuperación, neutralización e incineración controlada, indicando las condiciones en que debe realizarse.
Asimismo, deberá detallarse la depuración de aguas y los métodos recomendados para la manipulación, almacenamiento, transporte y respuesta ante incendios o derrames, incluyendo información sobre los productos de reacción y gases de combustión generados en caso de incendio. Finalmente, se deberá proporcionar información sobre los equipos de protección individual adecuados para minimizar los riesgos asociados a la exposición a la sustancia activa.
j.- EVALUACIÓN TOXICOLÓGICA
Para la identificación de peligros, deberá determinarse el Nivel de Efecto Adverso No Observable (NOAEL) con base en los estudios más representativos de los escenarios de uso del producto en Argentina, tanto para la evaluación de riesgos ocupacionales como dietarios. Se considerará el estudio toxicológico crónico más sensible y representativo de la vía de exposición evaluada.
En la evaluación de riesgo dietario, deberá establecerse la Ingesta Diaria Admisible (IDA) con base en el NOAEL del estudio toxicológico de largo plazo más sensible y representativo por vía oral, aplicando un factor de incertidumbre técnicamente justificado. Además, salvo prueba científica en contrario, deberá definirse una Dosis de Referencia Aguda (ARfD) basada en estudios de corto plazo por vía oral.
Asimismo, deberá realizarse la clasificación de peligros a la salud y de los peligros físicos de la sustancia activa conforme a la versión adoptada por la Autoridad Competente del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA) de las Naciones Unidas.
Definición del Residuo en Material Tratado:
Deberá proporcionarse la definición de residuos con fines de análisis de riesgo y monitoreo.
La inclusión de metabolitos o productos de degradación en la definición para evaluación del riesgo dependerá de su perfil toxicológico y su magnitud, cuando superen el DIEZ POR CIENTO (10 %) del total de residuos radioactivos.
Para la definición de residuos con fines de monitoreo, debe ser inequívoca y simple, preferentemente basada en un único compuesto (generalmente el compuesto parental), para todas las commodities.
Evaluación de la Exposición y Caracterización del Riesgo Dietario:
La evaluación del riesgo dietario, tanto crónico como agudo, se realizará conforme a metodologías internacionalmente aceptadas, considerando los ensayos de residuos supervisados para la sustancia activa, los valores de referencia toxicológicos (IDA y ARfD) y las estadísticas de consumo de la población argentina o una dieta representativa de la región.
Se considerará aceptable la exposición alimentaria crónica cuando la Ingesta Diaria Estimada Nacional sea inferior al CIEN POR CIENTO (100 %) de la IDA. De igual manera, la exposición alimentaria aguda será aceptable si la Ingesta Nacional Estimada de corto plazo es menor al CIEN POR CIENTO (100 %) de la ARfD.
Evaluación de la Exposición y Caracterización del Riesgo Laboral:
La evaluación de la exposición podrá basarse en estudios específicos o estimaciones mediante datos genéricos y modelos de simulación aceptados internacionalmente. Los escenarios de exposición deberán considerar las prácticas laborales, el uso y disponibilidad del equipo de protección personal, en concordancia con la práctica agrícola recomendada en la etiqueta.
El criterio de aceptabilidad del riesgo estará determinado por el Margen de Exposición (MOE), calculado como el cociente entre el NOAEL y la exposición estimada. Un MOE superior a CIEN (100) se considerará como riesgo aceptable.
En aquellos casos en que el valor de CIEN (100) pueda variar asociado a razones como relevancia de un estudio de ruta específica, ausencia de NOAEL u otras causas atendibles y válidas, deberá justificarse y validarse científicamente si se utiliza otro valor como límite de preocupación diferente a CIEN (100).
Período de Reingreso al Lote Tratado:
Cuando corresponda, se deberá realizar una evaluación de riesgo para el trabajador de reingreso al lote tratado. Para su determinación, deberán presentarse datos sobre toxicidad aguda dérmica, potencial irritante dérmico y ocular, y sensibilización dérmica del principio activo o del producto formulado.
VII - EFECTOS TÓXICOS SOBRE OTRAS ESPECIES (ÚNICAMENTE REQUERIDO PARA SUSTANCIAS ACTIVAS DE GRADO TÉCNICO NUEVAS).
Los estudios deberán realizarse conforme a las Test Guidelines y Guidance Documents de la OCDE u otras referencias internacionalmente reconocidas.
Para evaluar los efectos en aves, se deberán realizar estudios de toxicidad oral aguda, toxicidad dietaria subcrónica y efectos en la reproducción, utilizando especies validadas como faisán, codorniz o pato silvestre.
En cuanto a peces, se requerirá la evaluación de toxicidad aguda y crónica en especies como trucha arco iris o carpa, así como estudios de bioacumulación, los cuales serán exigidos de manera condicional para sustancias con un Log Kow superior a TRES (3).
Para invertebrados acuáticos, se deberá evaluar la toxicidad aguda y crónica en Daphnia magna. Asimismo, se evaluarán los efectos en organismos acuáticos mediante estudios sobre el crecimiento de algas, utilizando Selenastrum capricornutum u otra especie validada, y la inhibición del crecimiento de plantas acuáticas, tomando como referencia Lemna sp.
El impacto sobre invertebrados terrestres deberá analizarse mediante estudios de toxicidad aguda y crónica en abejas. Para las abejas adultas, se evaluará la toxicidad oral y por contacto (DL50); mientras que la toxicidad oral aguda en larvas y los estudios crónicos en adultos y larvas se determinará será requerido condicionalmente cuando la sustancia activa sea sistémica en el vegetal y presente un DL50 inferior a ONCE (11) miligramos/abeja. Quedan exceptuados los formulados de uso exclusivo en poscosecha, invernáculos o cebos.
Además, se deberá evaluar la toxicidad aguda en artrópodos benéficos en especies validadas como Aphidius rhopalosiphi, Chrysoperla carnea u Orius laevigatus. Para lombrices de tierra, se evaluará la toxicidad aguda y el impacto en la reproducción en Eisetia foetida u otra especie validada.
Asimismo, se analizarán los efectos sobre colémbolos en el suelo, particularmente en Folsomia candida y Folsomia fimetaria, mediante estudios de reproducción.
Finalmente, se evaluará la toxicidad en microorganismos del suelo, enfocándose en aquellos involucrados en procesos de nitrificación.
VIII - EFECTOS SOBRE EL MEDIO ABIÓTICO (ÚNICAMENTE REQUERIDO PARA SUSTANCIAS ACTIVAS DE GRADO TÉCNICO NUEVAS).
Comportamiento en el suelo.
Se requerirán estudios en al menos TRES (3) tipos de suelos patrón. Las evaluaciones deben conducirse considerando concentraciones de la sustancia equivalentes a la práctica agrícola crítica que se quiere registrar. Los suelos patrón deben ser representativos de los suelos agrícolas, pueden incluir uno de textura gruesa, otro de textura franca y otro de textura fina.
Se debe determinar la tasa de degradación, hasta el NOVENTA POR CIENTO (90%), y vías de degradación incluida la identificación de: Procesos que intervienen; Metabolitos y productos de degradación (Degradación Aeróbica y Anaeróbica/ Fotólisis); Absorción y desorción y movilidad de la sustancia activa y si es relevante, de sus metabolitos; Magnitud y naturaleza de los residuos remanentes.
Comportamiento en el agua y en el aire.
Se debe determinar la tasa de degradación, hasta el NOVENTA POR CIENTO (90%), y vías de degradación. Biodegradación.
m - EVALUACIÓN DE RIESGO AMBIENTAL
Formulación del problema:
En función del patrón de uso y las formulaciones representativas a registrar, se deben determinar grupos potenciales de especies no blanco que podrían estar expuestas y requerir evaluación de riesgo.
Evaluación de la exposición ambiental
El destino ambiental de una sustancia química dependerá del patrón de uso definido en la formulación del problema, por ende se deben considerar tanto los factores, como el método de aplicación, los cultivos objetivo, la época del año en que se propone la aplicación de la sustancia activa y el área geográfica en la que se utilizará.
Como producto final se deberá definir la concentración ambiental estimada (CAE), definida como la cantidad de sustancia activa y sus metabolitos toxicológicamente relevantes en agua, suelo o sedimento y aire, como resultado del uso propuesto. En ausencia de modelos de exposición locales, se podrán utilizar los procedimientos desarrollados por la Agencia de Protección Ambiental de Estados Unidos (US-EPA).
La evaluación de riesgos ambientales será desarrollada como un proceso escalonado, de diferentes fases de refinamiento de la información, tendiente a reducir la incertidumbre en la toma de decisiones.
Para nuevas sustancias activas, las fases serán:
Fase 1: Estudios de laboratorio estándar requeridos en esta norma: propiedades fisicoquímicas, destino ambiental, toxicidad aguda y/o crónica. En esta fase de la evaluación de riesgos, se utiliza el llamado enfoque de 'peor caso', el cual combina los valores más altos de exposición (máxima biodisponibilidad), con los niveles más altos de toxicidad (especie sensible).
Fase 2: Modelos de estimación de la distribución y degradación ambiental del compuesto parental y sus metabolitos toxicológicamente relevantes en suelo, agua y aire. La elección estará determinada por las propiedades individuales y los usos de una sustancia. En ausencia de modelos de exposición locales, se deberán utilizar los procedimientos de estimación ambiental predicha desarrollados por la Agencia de Protección Ambiental de Estados Unidos (US-EPA).
Fase 3: Estudios de semi-campo o de campo simulados, en caso de que el riesgo de un producto no pueda evaluarse suficientemente luego de las fases de evaluación 1 y 2, particularmente para sustancias que son relativamente persistentes o exhiben una alta movilidad. Se deberá acordar con la Autoridad Competente el protocolo a utilizar.
Caracterización del riesgo
Como procedimiento para integrar los resultados del análisis de la exposición con los efectos ecotoxicológicos adversos potenciales, puede utilizarse el método determinístico que utilice la aproximación de los COCIENTES DE RIESGO (RQ), calculados como el cociente entre los CAE y los valores de ecotoxicidad aguda o crónica, según corresponda. Sin embargo, pueden aplicarse otras metodologías aceptadas internacionalmente, a fin de establecer perfil ecotoxicológico de la sustancia en estudio.
DECLARACION DE NUEVOS ESTABLECIMIENTOS ELABORADORES
La solicitud de registro de una sustancia activa grado técnico registrada previamente por el solicitante, proveniente de un nuevo establecimiento elaborador debe realizarse mediante la presentación de la información y documentación requerida en el presente Anexo, Título “III. INFORMACIÓN CONFIDENCIAL (para sustancias activas nuevas y equivalentes)”.
La Autoridad Competente puede dar curso a la gestión de la solicitud, aprobación y registro una vez admitida la presentación de la información y documentación requerida.
B - PRODUCTOS FORMULADOS:
Además de los productos formulados de síntesis este registro también contempla los siguientes supuestos:
Los formulados que entre sus componentes contengan productos sintéticos solos o en mezcla con sustancias de origen biológico que por su forma de uso generen una respuesta en la conducta o en la fisiología del organismo plaga que se desee controlar, de aquí en adelante serán llamados semioquímicos.
Los formulados que contengan bactericidas, fungicidas y/o insecticidas destinados a prevenir, detener y/o eliminar el ataque de bacterias, hongos y/o insectos que afecten a las maderas en sus distintas formas y serán denominados de aquí en adelante preservadores para la madera.
Los formulados que sean producidos en base a agentes microbianos de ocurrencia natural, o introducidos en el ambiente, para la prevención y/o el control de organismos considerados plaga de la agricultura; sustancias bioquímicas (extractos de origen vegetal, animal o microbiológico). Invertebrados para el control biológico de plagas de la agricultura.
En los casos que se desee inscribir un producto formulado, se deberá presentar la documentación descrita a continuación. dada la diversidad de características asociadas al origen, naturaleza y aptitud de uso, los y preservadores de madera serán tratados en el apartado de LOS REQUISITOS Y PROCEDIMIENTOS DE PRODUCTOS DE TRATO DIFERENCIADO (PTD).
I - INFORMACIÓN ADMINISTRATIVA
Formulario de solicitud de registro dependiendo el tipo de formulado firmado por el Apoderado o Representante Legal y el Responsable Técnico con incumbencia profesional en la materia.
Proyecto de marbete según normativa vigente.
Hoja de Datos de Seguridad, de acuerdo a la versión del Manual de las Naciones Unidas del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA), adoptada en la presente norma.
Patrones: en caso de que el SENASA así lo requiera, a los fines de control y vigilancia, la empresa registrante deberá aportar los patrones y metodología analítica de la/s sustancia/s activa/s, en las condiciones que establezca la Dirección General de Laboratorios y Control Técnico (DGLCyT) del SENASA.
Sistema de gestión de envases vacíos aplicado en cumplimiento de la Ley vigente.
Formulario II (condicionalmente requerido).
Autorización a terceros a hacer uso del registro de un principio activo grado técnico para el registro de un producto formulado.
II - INFORMACIÓN CONFIDENCIAL
a.- Certificado de Origen del producto formulado.
El Certificado de Origen debe ser original y emitido por el establecimiento elaborador.
Para establecimientos no ubicados en la REPÚBLICA ARGENTINA, para cada uno de los orígenes a registrar.
Debe incluir:
Identificación de la/s sustancia/s activa/s; Concentración declarada para cada sustancia activa Tipo de formulación armonizada; Nombre y domicilio real del establecimiento formulador.
b. - Declaración de composición cuali-cuantitativa del producto formulado firmada por el establecimiento elaborador o por la empresa registrante con carácter de declaración jurada y que deberá contener:
Nombre químico según criterios internacionales (IUPAC o CAS) y número CAS (cuando está disponible) de la/s sustancia/s activa/s y demás componentes.
Contenido nominal de la(s) sustancia(s) activa(s), en base CIEN POR CIENTO (100 %) de pureza, expresado en porcentaje en masa (% p/p) para los formulados sólidos o gaseosos, y en porcentaje masa/volumen (% p/v) para los formulados líquidos.
Contenido nominal, naturaleza química y función específica de cada uno de los componentes restantes incluidos en la formulación.
Tipo de formulación armonizada de acuerdo con la Norma IRAM 12074.
Se presentará una sola declaración de composición cuali-cuantitativa representativa para todas las plantas para las que se solicita el registro en la que debe figurar la razón social y dirección real de cada uno de los establecimientos fabricantes.
c. - Estudio de UN (1) lote
Se debe presentar el análisis de una muestra representativa de UN (1) lote de formulación para la cuantificación de la(s) sustancia(s) activa(s), proveniente de una de las plantas para la que se solicita el registro o del Laboratorio de desarrollo. Los valores de concentración informados para la(s) sustancia(s) activa(s) deberán cumplir con los límites establecidos en la Norma IRAM 12054, en función del contenido nominal declarado.
En caso de que el sponsor del estudio no coincida con la empresa registrante se deberá remitir una autorización del sponsor para la utilización de la información.
Deberá presentarse el estudio completo, de acuerdo con los principios de Buenas Prácticas de Laboratorio, convenientemente firmado y fechado.
Se establecen los siguientes contenidos mínimos para el reporte del estudio de 1 lote:
Índice de contenidos
Nombre y domicilio real del establecimiento elaborador Cuadro de resumen de resultados
Métodos de análisis, que incluya todas las ecuaciones necesarias para reproducir los cálculos realizados
Resultados
En caso de usar curvas de calibración incluir los gráficos identificando título y ejes (con magnitud y unidades). Además debe incluir la ecuación de la recta. Cuando corresponda incluir los cálculos de densidad. Se deben incluir en formato de cuadro los datos necesarios para poder reproducir los cálculos realizados.
Los cromatogramas representativos del lote y del patrón analítico, con encabezado (con fecha, hora, descripción) y reporte (áreas y tiempo de retención). Certificados de análisis de todos los patrones utilizados. Cuando sean generados por el fabricante o el laboratorio deben incluir el reporte de cuantificación e identificación. En caso de patrones secundarios incluir la cuantificación contra el patrón primario junto a su certificado de análisis. Certificado de análisis del lote analizado emitido y firmado por el formulador que incluya nombre y dirección del establecimiento formulador y fecha de elaboración y vencimiento del lote. De no disponer en el certificado de análisis la dirección del establecimiento elaborador, se debe presentar una declaración jurada indicando dicha información acompañando el certificado. Constancia BPL OECD del Laboratorio
Acreditación BPL del estudio.
b.- Proceso de formulación
Para cada proceso de formulación debe proveerse la siguiente información emitida y firmada por el fabricante o la empresa registrante:
Nombre y domicilio real de los establecimientos formuladores que interviene en el proceso. Caracterización general del proceso: deberá consistir en una descripción completa y detallada de cada una de las etapas que constituyen el proceso, especificando qué componentes son agregados en cada una y qué operación se realiza.
Identificación de los ingredientes usados para formular el producto.
Descripción de los equipos usados, con sus especificaciones.
Descripción de las condiciones que se controlan en cada etapa del proceso y los parámetros de control de calidad del producto terminado.
III - CUERPO TÉCNICO
Este cuerpo técnico se presentará con la documentación dispuesta por temas, a saber:
a- COMPOSICIÓN
Contenido de sustancia(s) activa(s), grado técnico, expresado en % p/p o % p/v según corresponda Métodos de análisis para la determinación del contenido de la(s) sustancia(s) activa(s).
Para el caso de preservadores de la madera: deberá expresarse en óxidos cualesquiera fueran las sustancias que los integran, (como lo establece la norma IRAM). Y adicionalmente mencionar el método de aplicación: pincelado, aspersión, inmersión, prolongada, momentánea, breve, inyección, baño caliente-frío.
b - PROPIEDADES DEL PRODUCTO FORMULADO
Propiedades Físicas y Químicas.
Aspecto: Tipo de Formulación Armonizada; Color
Estabilidad en el almacenamiento a baja temperatura y acelerada a alta temperatura
Densidad relativa Inflamabilidad
Acidez/Alcalinidad y potencial de hidrógeno (pH).
Propiedades físicas relacionadas con su uso.
Humectabilidad: Para polvos dispersables o mojables.
Persistencia de la espuma: Para formulados que se aplican con agua. Suspensibilidad: Para gránulos dispersables (WG), polvos mojables: (WP), suspensiones concentradas: (SC), Suspensión de encapsulado (CS) Análisis granulométrico en húmedo: Para los polvos mojables, las suspensiones concentradas (SC, FS), Suspoemulsión (SE).
Análisis granulométrico en seco: Para gránulos y polvos. Estabilidad de la emulsión: Para concentrados emulsionables. Corrosividad.
Incompatibilidad con otros productos: Con otro fitosanitario y/o fertilizante. Densidad: Para sólidos y líquidos.
Punto de inflamación: Para líquidos.
Viscosidad: Para aceites, suspensiones y concentrados emulsionables. Índice de sulfonación: (Residuo no Sulfonable) Para aceites y aceites emulsionables destinados a frutales u ornamentales).
Dispersión: Para gránulos dispersables.
Desprendimiento de gases: Para gránulos generadores de gas. Soltura o fluidez: Para polvos secos.
Índice de iodo: Índice de Iodo y de Saponificación. Sólo para aceites vegetales, no para losaceites minerales.
Velocidad de liberación. Para suspensiones acuosas encapsuladas (CS) y para la formulación mixta (ZC), mezcla de suspensiones encapsuladas y suspensiones concentradas (CS + SC).
Presión de vapor, si corresponde en función de la volatilidad.
IV. ENSAYOS DE EFICACIA AGRONÓMICA Y FITOTOXICIDAD
Aquellas recomendaciones de usos que no tengan antecedentes en Argentina o las modificaciones de los usos críticos ya autorizados, deberán respaldar la solicitud mediante informes de los ensayos de eficacia agronómica y fitotoxicidad.
V ETIQUETADO
El etiquetado deberá ajustarse a la normativa vigente, y conforme a la versión del Manual de las Naciones Unidas del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA) adoptada por la Autoridad Competente del Registro Nacional.
VI. ENVASES Y EMBALAJES PROPUESTOS.
a.- Envases.
Tipo(s)
Material(es)
Capacidad(es)
b.-Embalajes.
Tipo
Material
Procedimientos para la descontaminación y destino final de los embalajes.
VII - EFECTOS TÓXICOS EN ESPECIES MAMÍFERAS
En el caso que la empresa registrante no disponga de estudios toxicológicos in vivo, como alternativa de reemplazo, reducción o refinamiento de las pruebas con animales requeridas en este apartado, podrán presentarse estudios in vitro, siempre y cuando, dichos protocolos y ensayos hayan sido reconocidos por autoridades regulatorias internacionales para el Registro Nacional de Productos Fitosanitarios. En tales casos, el registrante deberá solicitar mediante una nota la aceptación de dichos estudios, mediante una justificación técnico-científica de su uso.
Se aplicarán los criterios de las directrices de la OCDE para justificar la no conducción de estudios in vivo.
a.- Toxicidad aguda.
Oral. Toxicidad oral aguda (DL50 oral)
Este estudio se requerirá en todos los casos excepto si el producto formulado es un gas o es altamente volátil.
Dérmica. Toxicidad cutánea aguda (DL50 cutánea)
Este estudio se requerirá a menos que:
El producto formulado es un gas o es altamente volátil. El producto formulado es corrosivo para la piel o presenta un pH menor a DOS (2) o mayor a ONCE COMA CINCO (11,5).
Inhalatoria. Toxicidad aguda por inhalación (LC50 inhalación) Este estudio se requerirá cuando el producto formulado es un gas o es altamente volátil; o consiste en, o en las condiciones de uso darán como resultado, un material respirable (por ejemplo, gas, vapor, aerosol o partículas).
b.- Irritación cutánea y ocular.
Irritación cutánea.
El estudio in vivo no debería realizarse si: El producto formulado es un gas o es altamente volátil. El producto formulado es corrosivo para la piel o presenta un pH menor a DOS (2) o mayor a ONCE COMA CINCO (11,5)
Existe información disponible que indica que satisface los criterios para ser clasificada como corrosiva para la piel o irritante ocular La sustancia es clasificada como muy tóxica por vía dermal
Irritación ocular.
Este estudio se requerirá a menos que:
El producto formulado sea corrosivo o severo irritante para la piel,
Presente un pH menor a DOS (2) o mayor a ONCE COMA CINCO (11,5)
La sustancia es clasificada como muy tóxica por vía dermal.
c.- Sensibilización cutánea.
Este estudio se requerirá a menos que el principio activo sea un sensibilizante conocido.
VIII - INFORMACIÓN MÉDICA OBLIGATORIA
Diagnóstico y síntomas de intoxicación
Tratamientos propuestos
Primeros auxilios. Antídotos
Tratamiento médico.
IX - DATOS DE LOS EFECTOS SOBRE EL AMBIENTE
a.- Toxicidad aguda en aves Estos estudios serán requeridos para todos los productos cuyos usos propuestos son en lugares abiertos. Toxicidad oral aguda (en faisán, codorniz, pato silvestre y otra especie validada).
b.- Toxicidad aguda sobre organismos acuáticos Estos estudios serán requeridos para todos los productos cuyos usos propuestos son en lugares abiertos. Concentración letal media de NOVENTA Y SEIS HORAS (96 h) en peces (en trucha arco iris, carpa y otras especies validadas).
d.- Efectos tóxicos en polinizadores e invertebrados no blanco
Este estudio será requerido si por el uso propuesto pudiere resultar una exposición de las abejas.
Toxicidad oral aguda para las abejas adultas (DL50 oral adultas).
Toxicidad por contacto agudo para las abejas adultas (DL50 contacto adulto).
X EVALUACIÓN TOXICOLÓGICA Y ECOTOXICOLÓGICA
Clasificación toxicológica y ecotoxicológica para el etiquetado del producto fitosanitario formulado y la Hoja de Datos de Seguridad, de acuerdo a la versión del Manual de las Naciones Unidas del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA), adoptada por la Autoridad Competente del Registro Nacional.
Se considerarán como excepción las clasificaciones de peligro de aves y abejas, las cuales adoptarán las clasificaciones de la USEPA.
XI - INFORMACIÓN SOBRE EL MANEJO DE DESECHOS DEL PRODUCTO FORMULADO
a.- Métodos de disposición final de los desechos Se debe incluir el o los procedimientos más adecuados para eliminación de remanentes o residuos de las aplicaciones, así como de los envases.
b.- Procedimientos para la destrucción del producto formulado y para la descontaminación.
Se deben presentar los procedimientos y métodos a seguir para la destrucción o inactivación del producto formulado.
Incineración controlada (condiciones).
Posibilidades de neutralización.
c.-Procedimientos de recuperación en caso de derrame Detallar el o los procedimientos para la recuperación del producto formulado.
d.- Depuración de las aguas Detallar el o los procedimientos a seguir para la depuración de fuentes de agua contaminadas con el producto formulado.
e.- En caso de incendio Identificar los productos de reacción y gases de combustión poniendo énfasis en aquellas sustancias que presentan riesgo toxicológico humano o ambiental y con base en lo anterior establecer y presentar un protocolo de respuesta ante una emergencia específica.
XII - EVALUACIÓN DE RIESGO
La Autoridad Competente establecerá qué categoría de productos deberán presentar una evaluación de riesgo en cumplimiento con lo establecido en el Anexo VII en el que se detalla el análisis de riesgos.
DECLARACIÓN DE NUEVOS ESTABLECIMIENTOS ELABORADORES DE PRODUCTO FORMULADO REGISTRADO
Se presentará una sola declaración de composición cuali-cuantitativa representativa para todas las plantas para las que se solicita el registro, si el producto a registrar no presenta diferencias entre las mismas, lo que deberá indicarse bajo declaración jurada.
A tal efecto, debe presentar para cada nuevo establecimiento:
1) Declaración jurada
Formulario de declaración jurada indicando la razón social y dirección real del establecimiento o planta a través de la plataforma SIGTrámites del Senasa o por aquellos medios que el Senasa disponga a tal fin, firmada por el Apoderado o Representante Legal.
* En virtud que por declaración jurada del responsable titular del registro, no existen diferencias significativas entre los productos obtenidos en los diferentes establecimientos o plantas de un mismo productos formulado, de observarse incumplimiento por acciones de control sobre el productos formulado de un establecimiento o planta, las medidas que la Autoridad Competente adopte serán aplicadas a todos los establecimientos o plantas declaradas para el registro involucrado.
2) Certificado de Origen del producto formulado.
El Certificado de Origen debe ser original y emitido por el fabricante y estar debidamente legalizado por las autoridades del país de origen.
Debe incluir:
• identificación de la sustancia activa.
• contenido mínimo declarado en % p/p.
• Denominación/razón social y domicilio real del establecimiento elaborador.
• (*)Nombre y dirección de la empresa que registrará el producto ante SENASA.
En caso de que no figure esta información se deberá remitir una nota emitida por el establecimiento elaborador donde conste la autorización para realizar el registro del producto en el país.
NUEVA MARCA COMERCIAL PARA UN PRODUCTO FORMULADO YA REGISTRADO
Si un registrante solicita un registro de un producto ya registrado por el mismo solamente se debe mencionar expediente aprobado.
Si el registro corresponde a otra persona física o jurídica, deberá presentarse la autorización de uso correspondiente.
I. Información legal y administrativa.
• Proyecto de marbete según normativa vigente.
• Hoja de Datos de Seguridad, de acuerdo a la versión del manual de las Naciones de las Naciones Unidas del “Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos”, adoptada por la Autoridad Competente del Registro Nacional de Terapéutica Vegetal..
Formulario II (condicionalmente requerido). Autorización a terceros de hacer uso del registro de un principio activo grado técnico para el registro de un producto formulado.
Requisitos y procedimientos DE COADYUVANTES DE APLICACIÓN.
I. Información legal y administrativa.
• Formulario de solicitud del producto formulado con aptitud COADYUVANTE,
• Proyecto de marbete según normativa vigente.
• Hoja de Datos de Seguridad, de acuerdo a la versión del manual de las Naciones Unidas del “Sistema Globalmente Armonizado de Etiquetado de Productos Químicos”, adoptada por la Autoridad Registro Nacional de Terapéutica Vegetal.
• Sistema de gestión de envases vacíos aplicado en de la Ley vigente.
II. Cuerpo técnico
1. Certificado de Origen del producto formulado.
El Certificado de Origen emitido por el fabricante (*) para cada uno de los orígenes a registrar, debe incluir:
• identificación de la sustancia principal que se pretende registrar como coadyuvante.
• contenido mínimo declarado.
• tipo de formulación
• Denominación/razón social y domicilio real del establecimiento fabricante.
(**)Nombre y dirección de la empresa que registrará el producto ante SENASA.
(*) Plantas del extranjero debidamente legalizado por las autoridades del país de origen.
(**) En caso de que no figure esta información se deberá remitir una nota emitida por el establecimiento fabricante donde conste la autorización para realizar el registro del producto en el país
2. Declaración de composición cuali-cuantitativa del producto formulado
La misma debe estar firmada por el establecimiento fabricante o por la empresa registrante con carácter de declaración jurada:
• Nombre químico según criterios internacionales (IUPAC o CAS) y número CAS (cuando esté disponible) de la/s sustancia/s activa/s y demás componentes.
• Contenido nominal de la(s) sustancia(s) a registrar con clase de uso coadyuvante, en base 100 % de pureza, expresado en porcentaje en masa (% p/p) para los formulados sólidos o gaseosos, y en porcentaje masa/volumen (% p/v) para los formulados líquidos.
• Contenido nominal, naturaleza química y función específica de cada uno de los componentes restantes incluidos en la formulación.
• Tipo de formulación armonizada de acuerdo con la Norma IRAM 12074.
• Método de análisis.
Se presentará una sola declaración de composición cuali-cuantitativa representativa para todas las plantas para las que se solicita el registro en la que debe figurar la razón social y dirección real de cada uno de los establecimientos fabricantes.
3. Información toxicológica y ecotoxicológica
El Dossier debe contener información científica respaldatoria con la caracterización toxicológica y/o ecotoxicológica para la clasificación de los productos formulados en función de la información disponibles para clasificar los peligros de los componentes principales y los co-formulantes, siendo posible aplicar los principios de extrapolación y estimación de la toxicidad aguda (ETA), según el manual de las Naciones Unidas del “Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos’’.
Requisitos y procedimiento DE SEMIOQUÍMICOS EN BASE A SUSTANCIAS ACTIVAS SINTÉTICAS.
Requisitos generales de registro de productos formulados que entre sus componentes contengan productos sintéticos, solos o mezcla con sustancias de origen biológico, que por su forma de uso se espera generen una respuesta en la conducta o en la fisiología de organismo plaga que se pretende controlar o monitorear. Comprenden a:
• Dispositivos de captura, cuando el uso del semioquímico tenga como objetivo la atracción sexual o alimentaria de plagas, con el objetivo de monitorear poblaciones de una especie.
• Atracticidas, cuando mediante el uso de cebos o de dispositivos de captura masivos, se combina el uso de semioquímicos (como atrayente sexual o alimentario), con el empleo de insecticidas o mecanismos de captura física para disminuir la abundancia y daños de las plagas en los cultivos.
• Confusión Sexual, cuando mediante el asperjado o el uso masivo de emisores de semioquímicos se busca la saturación del medioambiente con una feromona sexual con el fin de interferir en comportamiento de apareamiento de la plaga, para disminuir la abundancia y daños de las plagas en los cultivos.
• Repelentes, cuando mediante el asperjado o el uso masivo de emisores se busca la repelencia de plagas a fin de disminuir los daños de las mismas en los cultivos.
I. Información legal y administrativa.
• Formulario de solicitud del producto formulado con aptitud SEMIOQUÍMICO,
• Patrones: en caso de que la autoridad de aplicación así lo requiera, a los fines de control y vigilancia, la empresa registrante deberá aportar los patrones y metodología analítica de la/s sustancia/s activa/s, en las condiciones que establezca la Dirección General de Laboratorios y Control Técnico (DGLCyT) del SENASA.
• Proyecto de marbete según normativa vigente
• Hoja de Datos de Seguridad, de acuerdo a la versión del manual de las Naciones Unidas del “Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos’”, adoptada por la Autoridad Competente del Registro Nacional de Terapéutica Vegetal.
• Sistema de gestión de envases vacíos aplicado en cumplimiento de la Ley vigente, si corresponde.
II. INFORMACIÓN TÉCNICA.
NOTA: los productos formulados utilizados exclusivamente para Monitoreo de Plagas Agrícolas se encuentran exentos de presentación de información técnica respaldatoria
• Certificado de Origen de la Sustancia activa grado técnico.
El Certificado de Origen emitido por el fabricante (4), debe incluir:
• identificación de la sustancia activa.
• contenido mínimo declarado.
• Denominación/razón social y domicilio real del establecimiento fabricante.
(**) Nombre y dirección de la empresa que registrará el producto ante SENASA.
(*) Establecimiento del extranjero debidamente legalizado por las autoridades del país de origen.
(**) En caso de que no figure esta información se deberá remitir una nota emitida por el establecimiento fabricante donde conste la autorización para realizar el registro del producto en el país.
• Certificado de Origen del producto formulado.
El Certificado de Origen emitido por el fabricante (*) para cada uno de los orígenes a registrar, debe incluir:
• identificación de la/s sustancia/s activa/s.
• contenido mínimo de SUSTANCIA ACTIVA declarado.
• tipo de formulación
• Denominación/razón social y domicilio real del establecimiento fabricante.
• (**) Nombre y dirección de la empresa que registrará el producto ante SENASA.
(*) Establecimiento del extranjero debidamente legalizado por las autoridades del país de origen.
(**) En caso de que no figure esta información se deberá remitir una nota emitida por el establecimiento fabricante donde conste la autorización para realizar el registro del producto en el país.
• Declaración de composición cuali-cuantitativa del producto formulado
La misma debe estar firmada por el responsable del establecimiento fabricante o de la empresa registrante con carácter de declaración jurada:
• Nombre químico según criterios internacionales (IUPAC o CAS) y número CAS (cuando esté disponible) de la/s sustancia/s activa/s y demás componentes.
• Contenido nominal de la(s) sustancia(s) activa(s), en base 100 % de pureza, expresado en porcentaje en masa (% p/p) para los formulados sólidos o gaseosos, y en porcentaje masa/volumen (% p/v) para los formulados líquidos.
• Contenido nominal, naturaleza química y función específica de cada uno de los componentes restantes incluidos en la formulación.
• Tipo de formulación armonizada de acuerdo con la Norma IRAM 12074.
Se presentará una sola declaración de composición cuali-cuantitativa representativa para todas las plantas para las que se solicita el registro en la que debe figurar la razón social y dirección real de cada uno de los establecimientos fabricantes.
• Certificado de Análisis cuali-cuantitativo (condicionalmente requerido*)
Certificado de Análisis cuali-cuantitativo de una muestra representativa de UN (1) lote de formulación del producto formulado a registrar con nombre y firma del Responsable técnico del laboratorio que realizó el análisis.
• Propiedades físicas y químicas del producto formulado
Se requerirá la presentación de estudios para aquellos semioquímicos para los que se espera exposición (i.e. pulverizables), para los restantes se podrá presentar dossier sobre la base de la información técnica disponible para la/a sustancia/a activa/s y los coformulantes.
• Aspecto (estado físico y color):
• Densidad.
• pH.
• Estabilidad en el almacenamiento
• Tensión superficial (cuando corresponda)
• Solubilidad (cuando corresponda)
• Suspensibilidad (cuando corresponda)
• Ensayos de eficacia agronómica y fitotoxicidad (cuando corresponda).
En caso de no existir antecedentes de uso autorizado en Argentina, en relación a la naturaleza química del semioquímico (dosis, tipo de formulación/dispositivo o técnica de aplicación, etc), deben presentarse informes de los ensayos de eficacia agronómica, de acuerdo con lo establecido en el presente Manual según lo establecido en el Cap. 20 del presente manual.
• Información toxicológica y ecotoxicológica
Debe presentar un Dossier con información científica respaldatoria con la caracterización toxicológica para la clasificación de los productos formulados en función de la información disponible para clasificar los peligros de los ingredientes activos y los co-formulantes, siendo posible aplicar los principios de extrapolación y estimación de la toxicidad aguda (ETA), según el manual de las Naciones Unidas del “Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos”.
PRODUCTOS FORMULADOS A BASE DE AGENTES MICROBIANOS,
SUSTANCIAS BIOQUÍMICAS O INVERTEBRADOS
REQUISITOS GENERALES
a) Los Ensayos de eficacia agronómica deben ser realizados localmente, por especie plaga que se pretenda registrar, y cumplir con requisitos establecidos en la normativa vigente. Los ensayos de eficacia agronómica de un bioinsumo sin antecedentes de registro en el país, deben llevarse a cabo en al menos TRES (3) zonas agroecológicas distintas de la REPÚBLICA ARGENTINA, durante al menos DOS (2) campañas agrícolas. Podrán ser admitidos ensayos en una misma campaña siempre que la DAYB cerciore que los ensayos reflejen variabilidad climática, se realicen en al menos tres zonas agroecológicas y respeten el mínimo número de ensayos, SEIS (6).
b) Para bioinsumos a base de organismos modificados genéticamente, en todos casos deberá contar previamente al registro con la evaluación favorable realizada por la Coordinación de Innovación y Biotecnología perteneciente a la Dirección Nacional de Bioeconomía de SAGYP y la Comisión Nacional Asesora de Biotecnología Agropecuaria (CONABIA), conforme a lo establecido en las normativas vigentes. C) Cuando el producto a registrar proceda de la extracción de recursos natural nativo, la empresa registrante debe contar con el correspondiente permiso de colecta/acceso a los recursos genéticos.
INFORMACIÓN LEGAL Y ADMINISTRATIVA GENERAL.
Presentar de acuerdo con B - PRODUCTOS FORMULADOS - I. INFORMACION ADMINISTRATIVA.
Consideración para productos bioinsumos: Certificado de Origen firmado por personal responsable de la planta de producción / Establecimiento elaborador del Bioinsumo. En caso de provenir de una planta extranjera, deben estar debidamente autenticados y/o apostillados por las autoridades del país de origen.
INVERTEBRADOS PARA EL CONTROL BIOLÓGICO
I. Información legal y administrativa
• Acreditación del Registro de planta de cría.
• Certificado de origen del hospedero/presa que acompaña al invertebrado (cuando corresponda)
• Permiso de importación del invertebrado y del hospedero/presa que acompaña al invertebrado (cuando corresponda)
• Certificado de la Cuarentena emitido por el Laboratorio oficial (cuando corresponda)
• Autorización de colecta emitida por la autoridad de aplicación ambiental (cuando corresponda).
• Muestra de referencia para ser conservada en el laboratorio del SENASA.
II. Información técnica
A. Identidad y proceso de fabricación
• Nombre científico e identificación taxonómica (indique sinonimia, subespecie, variedad o raza si corresponde).
• Descripción morfológica.
• Método utilizado para identificar el invertebrado (taxonomía clásica, técnicas moleculares, etc.). Incluir claves de identificación morfológica cuando corresponda
• Estado de desarrollo biológico del invertebrado.
• Descripción detallada de los sustratos orgánicos e inorgánicos incluidos en el
• producto comercial.
• Nombre científico, características y función del hospedero/presa que acompaña al
• invertebrado (cuando corresponda).
• Libro de procedimiento y control de calidad para las etapas de crianza de los invertebrados.
B. Propiedades biológicas y utilización agronómica.
• Rango de hospederos/presas conocidos.
• Pruebas de especificidad, que indiquen cualquier riesgo potencial sobre especies no objetivos o para la salud de las personas y animales.
• Antecedentes de los resultados, de la introducción y/o liberación del organismo en otros países (para organismo exóticos).
• Estabilidad frente a diferentes condiciones climáticas
• Tipo de organismo (depredador, parasitoide o parásito), y estado o fase de desarrollo en el que será comercializado.
• Condiciones de liberación
• Método de liberación
• Dosis
• Intervalo entre liberaciones
• Periodo en que deben suspenderse las aplicaciones de otros productos fitosanitarios
• (antes y después de la liberación)
• Vida útil del producto
• Condiciones de almacenamiento, transporte y seguridad
• Informes de los ensayos de eficacia agronómica para los usos propuestos.
C. Envases y Embalajes según B - PRODUCTOS FORMULADOS - V. ENVASES Y EMBALAJES PROPUESTOS.
D. Medidas de Seguridad:
• Métodos recomendados y precauciones durante la multiplicación, fraccionamiento, almacenamiento, liberación y manipuleo general del producto. Indicar procedimientos de actuación en caso de accidentes.
Requisitos y procedimientos para SUSTANCIAS BIOQUIMICAS FORMULADAS A BASE DE EXTRACTOS DE ORIGEN VEGETAL, ANIMAL O MICROBIOLÓGICO.
I. Información legal y administrativa específica
a) Certificado de Composición original firmado por el Responsable Técnico de la empresa formuladora.
b) Informe de Análisis cuali-cuantitativo del producto formulado a registrar firmado por el Responsable técnico del laboratorio.
c) Autorización de colecta emitida por la autoridad de aplicación ambiental (cuando corresponda)
d) Acreditación del Registro de planta formuladora / establecimiento.
e) Sistema de gestión de envases vacíos aplicado en cumplimiento de la Ley vigente, si corresponde.
I. Información técnica
A. Información de la sustancia activa
a) Productor de la sustancia activa
b) Nombre común: Aceptado o propuesto por ISO y Sinónimo (si lo tiene).
c) Nomenclatura química (aceptado o propuesto por IUPAC), si corresponde.
d) Fórmula empírica, si corresponde.
e) Fórmula estructural (incluyendo estereoquímica de isómeros activos si corresponde), si corresponde.
f) Masa molecular, si corresponde.
g) Grupo químico al que pertenece.
h) Grado de pureza (contenido mínimo de sustancia activa en g/kg).
i) Caracterización de los otros componentes presentes en el extracto.
j) Caracterización de otros componentes agregados (ej. estabilizantes). Identificación y concentración.
B. Método de obtención de la sustancia activa
Esta información deberá venir respaldada por un Certificado emitido por el fabricante con el método de obtención, especificando los reactivos y solventes empleados en el proceso. Indicar los equipos utilizados y las condiciones de operación, especificando los parámetros controlados durante el proceso.
C. Métodos analíticos para la cuantificación y la identificación de la sustancia activa:
• El contenido mínimo declarado de la sustancia activa debe estar sustentado en el
• análisis de 5 lotes del grado técnico. Se deberá adjuntar la documentación de identificación y trazabilidad de los 5 lotes presentados
• Deberá presentarse el estudio completo de cuantificación de los 5 lotes, de acuerdo con los lineamientos de Buenas Prácticas de Laboratorio que defina la DGLyCT del Senasa, convenientemente firmado y fechado. El informe del laboratorio deberá incluir la descripción del método de ensayo, los registros cromatográficos, las tablas de cuantificación (incluyendo los datos crudos) y los Certificados de Análisis de los Patrones y la Muestra referidos en el estudio que deberán cumplir con protocolos internacionalmente reconocidos.
• Deberán facilitarse descripciones completas de la metodología que deberá estar convenientemente validada.
Deberán presentarse los parámetros de validación obtenidos:
• Linealidad.
• Límite de detección y cuantificación.
• Especificidad.
• Exactitud y repetibilidad.
Deberá realizarse la identificación de la sustancia activa en los 5 lotes por un método espectrométrico (MS, RMN, IR).
D. Propiedades físicas y químicas de la sustancia activa
a) Aspecto:
• Estado físico
• Color
b) Densidad
c) pH.
d) Estabilidad en el almacenamiento
B. Información del producto formulado
Identidad y proceso de fabricación del producto formulado
a) Declaración de la composición cuali-cuantitativa del producto formulado firmada por el Representante Legal con carácter de Declaración Jurada, que deberá incluir:
• Contenido de la(s) sustancia(s) activa(s), en base 100 % de pureza, expresado en porcentaje en masa (% p/p) para los formulados sólidos o gaseosos, y en porcentaje masa/volumen (% p/v) para los formulados líquidos.
• Contenido, naturaleza química y función específica de cada uno de los componentes restantes incluidos en la formulación.
• Tipo de formulación armonizada de acuerdo con la Norma IRAM 12074.
b) Análisis de una muestra representativa de UN (1) lote de formulación para la cuantificación de la(s) sustancia(s) activa(s). Deberá presentarse el estudio completo, de acuerdo con los lineamientos de Buenas Prácticas de Laboratorio que defina la DGLyCT del Senasa, convenientemente firmado y fechado,. El informe del laboratorio deberá incluir la descripción del método de ensayo, los registros cromatográficos, las tablas de cuantificación (incluyendo los datos crudos) y los Certificados de Análisis de los Patrones y la Muestra referidos en el estudio que deberán cumplir con protocolos internacionalmente reconocidos. Asimismo, se deberá adjuntar la documentación de identificación y trazabilidad de la muestra analizada.
Los valores de concentración informados para la(s) sustancia(s) activa(s) deberán cumplir con los límites establecidos en la Norma IRAM 12054, en función del contenido respectivo declarado.
c) Certificado de Origen del producto formulado, donde conste la identificación de la(s) sustancia(s) activa(s), su concentración declarada, el tipo de formulación armonizada y el nombre y la localización de la planta formuladora. El Certificado de Origen deberá estar debidamente legalizado por las autoridades del país de origen.
F. Descripción del proceso de formulación, que deberá contener la siguiente información:
a) Nombre y localización de la planta formuladora que interviene en el proceso.
b) Caracterización general del proceso: deberá consistir en una descripción completa y detallada de cada una de las etapas que constituyen el proceso, especificando qué componentes son agregados en cada una y qué operaciones se realizan.
c) Identificación de los ingredientes usados para formular el producto.
d) Descripción de los equipos usados, con sus especificaciones.
e) Descripción de las condiciones que se controlan en cada etapa del proceso y los parámetros de control de calidad del producto terminado.
f) Descripción de posibles reacciones posteriores al proceso de formulación que ocurran entre los ingredientes activos, o entre éstos y cualquier otro componente de la formulación o el envase; posible migración de materiales del envase al producto.
G. Propiedades físicas y químicas del producto formulado.
a) Aspecto:
• Estado físico.
• Color.
b) Densidad.
c) pH.
d) Estabilidad en el almacenamiento
e) Tensión superficial (cuando corresponda)
f) Suspensibilidad (cuando corresponda)
g) Solubilidad (cuando corresponda)
H. Información toxicológica, ecotoxicológica y ambiental
H.1. Toxicidad aguda en mamíferos.
a) Toxicidad oral aguda (DL50 oral)
b) Toxicidad cutánea aguda (DL50 cutánea)
c) Toxicidad aguda por inhalación (LC50 inhalación). Condicionalmente requerido
d) Irritación cutánea. Se deberá seguir un enfoque escalonado:
d.1. evaluación de la corrosividad dérmica utilizando un método de prueba in vitro validado;
d.2. un estudio de irritación dérmica inicial in vivo con un animal, y donde no se observan efectos adversos;
d.3. pruebas confirmatorias in vivo con uno o dos animales adicionales.
e) Irritación ocular. Se deberá seguir un enfoque escalonado:
e.1. el uso de una prueba de irritación / corrosión cutánea in vitro para predecir irritación / corrosión ocular;
e.2. un estudio inicial de irritación ocular in vivo con un animal, y donde no se observan efectos adversos;
e.3. pruebas confirmatorias in vivo con uno o dos animales adicionales.
f) Sensibilización cutánea.
Este estudio se requerirá a menos que el principio activo sea un sensibilizante conocido. Por ende, la no presentación de estudios de sensibilidad dermal debe ser documentada con un estudio puente donde se haya determinado que la sustancia es un sensibilizante dermal conocido.
g) Mutagenicidad:
Un estudio inicial de mutagenicidad es requerido mínimamente. Estudios subsiguientes pueden o no ser requeridos de acuerdo con los resultados obtenidos en los ensayos interiores.
Para cumplimentar este requisito se recomienda un enfoque por etapas: (IN VITRO)
g.1. Estudio de mutación genética en bacterias (por ej. Test de Ames)
g.2. Estudio de citogenética en células de mamíferos o test de micronúcleo. Este estudio no tiene que realizarse si hay datos negativos de estudios de citogenética in vivo.
g.3. Estudio de mutación genética en células de mamíferos, si se obtuvo un resultado negativo en g.1 y g.2., este estudio no será requerido.
g.4. Serán requeridos estudios de mutagenicidad in vivo en el caso de haber obtenido resultados positivos en los estudios in vitro.
H.2. Estudios subcrónicos y crónicos
Para nuevas sustancias sin antecedentes de registro en Argentina. Debe presentar un Dossier conteniendo información científica respaldatoria sobre la caracterización toxicológica, ecotoxicológica y destino ambiental. Esta información puede provenir de estudios toxicológicos y ecotoxicológicos realizados en laboratorio, mediante protocolos reconocidos internacionalmente, o de referencias bibliográficas completas procedentes de revistas indexadas. La evaluación de peligros se utilizará para cuantificar o desestimar los efectos potencialmente dañinos de la sustancia a ser comunicados en la etiqueta del producto y en la Hoja de datos de seguridad.
En aquellos casos, que por su forma de uso se espere exposición hacia el aplicador y el consumidor, debe presentar una Evaluación de la exposición y caracterización del riesgo dietario al consumidor, y una Evaluación de la exposición y caracterización del riesgo laboral
I. Ensayos de eficacia agronómica y fitotoxicidad.
Informes de los ensayos de eficacia agronómica de los usos propuestos.
J. Envases y embalajes propuestos según B -
PRODUCTOS FORMULADOS - V. ENVASES Y EMBALAJES PROPUESTOS.
AGENTES MICROBIANOS DE CONTROL BIOLÓGICO
Cada nuevo aislado se considerará como un nuevo ingrediente activo y debe registrarse independientemente de cualquier ingrediente activo registrado de manera similar. El nuevo aislamiento debe tener un identificador único después del nombre taxonómico del microorganismo. Esto no excluye la posibilidad de utilizar datos de otra cepa con antecedentes de registro, como estudios puente, para iniciar el proceso de registro.
Los productos elaborados en base a microorganismos sin antecedentes de registro en el país deberán realizar una solicitud de autorización de muestra y someterse a la evaluación de riesgo por parte autoridad competente en lo referente a los aspectos cuarentenarios y de impacto a nivel de los recursos naturales y/o de la salud. En caso de ser un microorganismo nativo, debe presentar la autorización de permiso de colecta de la provincia originaria.
Se considera como criterio excluyente de la presentación a registro los microorganismos que son patógenos primarios de animales, plantas y el ambiente. Aquellos que puedan presentar proximidad taxonómica con los microorganismos que son patógenos, deberán garantizar la inocuidad para la salud y el ambiente mediante los estudios que la avalen.
I. Información legal y administrativa
a) Certificado de Composición original con nombre y firmado por el Responsable Técnico de la empresa formuladora.
b) Informe de Análisis cuali-cuantitativo del producto formulado a registrar firmado por el Responsable técnico del laboratorio.
d) Acreditación del Registro de planta biológica / establecimiento elaborador.
e) Certificado de la Cuarentena emitido por el Laboratorio oficial (cuando corresponda)
II. Información técnica
A. Identidad y proceso de fabricación
a) Solicitante del registro.
b) Productor /proveedor de materia prima.
c) Nombre y descripción de la especie, caracterización de la cepa y origen. Taxonomía: la identificación deberá realizarse al menor nivel taxonómico en que se produzcan diferencias en el accionar del microorganismo o sus productos metabólicos (especie, subespecie, cepa, serotipo, patovar según corresponda al microorganismo), nivel de seguridad en la identificación por encima del 98%. Origen (nativo, exótico, OGM, etc).
d) Metodología de identificación y caracterización del microorganismo: Señalar la metodología y criterios utilizados para la identificación (morfología, bioquímica, serología, identificación molecular, etc.).
e) Datos de depósito de microorganismo: identidad de la colección y número de código de aislamiento.
f) Método de producción y control de calidad. Esta información deberá venir respaldada por una monografía, que contenga la descripción o flujograma del proceso productivo, especificando las condiciones, medios de cultivo, aditivos y otros empleados. Señalar las técnicas para garantizar la uniformidad y estandarización de la producción, y los procedimientos o metodología para el control de calidad.
g) Concentración del microorganismo (ingrediente activo) expresado en unidades infectivas conocidas. Declarar mínimo garantizado a la elaboración y al vencimiento, expresado en unidad de medida que corresponda.
h) Contenido e identidad de otros componentes. (Como condensados, medio de cultivo, promotores de la alimentación, entre otros). Señalar identidad y contenido máximo de microorganismos contaminantes, expresados en la unidad de medida que corresponda (CR).
i) Señalar identidad y caracterización de toxinas y metabolitos que forma el microorganismo, indicando, cuando corresponda, si tales toxinas o metabolitos ejercen o no la acción a la que está destinado al uso.
j) Estabilidad genética del agente de control biológico microbiano.
B. Métodos analíticos.
Indicar los métodos de análisis o técnicas, adjuntando su descripción, para:
a) Identificación del microorganismo al nivel establecido de especificidad (especie, subespecie, biotipo, cepa, etc.)
b) Determinación del contenido y viabilidad del microorganismo en el producto formulado.
C. Identidad del producto formulado.
a) Contenido del microorganismo: Indicar el contenido del o de los microorganismos en el producto formulado, indicando el contenido mínimo y nominal del material viable e inviable.
b) Co-formulantes: Indicar identidad, función y contenido máximo de aditivos, cuando corresponda; como los subproductos, condensados, medios de cultivo. Para la identificación de las sustancias químicas señalar nombre químico, nombre común, (si existe) y número CAS (si existe).
c) Certificado de origen y composición cuali-cuantitativo, emitido por el productor.
d) Propiedades físico químicas del producto formulado
• Color
• Estado físico
• pH
e) Estabilidad en diferentes condiciones ambientales (luz solar, pH, aire, temperatura, metales y sus iones).
f) Actividad acuosa (miscibilidad)
g) Estabilidad en el almacenamiento
h) Suspensibilidad (cuando corresponda)
C. Propiedades biológicas y utilización agronómica.
Informes de los ensayos de eficacia agronómica de los usos propuestos.
D. Evaluación tóxico-patológica. Efectos sobre la salud humana Debe presentar un Dossier con información científica respaldatoria con la caracterización toxico-patológica para la clasificación de los productos formulados en función de la información disponible para clasificar los peligros de los ingredientes activos y los co-formulantes. Mínimamente debe considerar información de toxicidad aguda/patogenicidad Oral aguda, Pulmonar aguda, Indicación de alergia/hipersensibilidad. Será condicionalmente exigido el cultivo de células (solo para virus). Para microorganismos sin antecedentes de uso en Argentina debe presentar información sobre la toxicidad/patogenicidad subcrónica
E. Evaluación tóxico-patológica. Efectos sobre otros organismos no objetivo
Debe presentar un Dossier con información científica respaldatoria con la caracterización ecotoxico-patológica para la clasificación de los productos formulados en función de la información disponible para clasificar los peligros de los ingredientes activos y los coformulantes en aves, peces de agua dulce y abejas.
F. Información respecto a la seguridad
Procedimientos para la destrucción del agente microbiológico, producto de su metabolismo, producto formulado, agentes biológicos mutantes, indicando las condiciones físicas o químicas específicas para obtener la desactivación o descomposición del material biológico/producto.
Métodos recomendados y precauciones de manejo durante la fabricación, formulación, almacenamiento, transporte, uso y manipuleo general del agente / producto. Información sobre equipos de protección personal (si corresponde). Procedimientos de limpieza y descontaminación de equipos de aplicación y áreas contaminadas.
G. Envases y embalajes propuestos según B - PRODUCTOS FORMULADOS - V. ENVASES Y EMBALAJES PROPUESTOS.
Consideraciones para preservadores de la madera: Los biocidas gaseosos para la fumigación de viviendas y artículos de madera como así de los destinados a reimpregnación de postes y los destinados a usos domisanitarios quedan excluidos de la obligatoriedad de inscribirse en el Registro Nacional.
PROCEDIMIENTO PARA LA EXTENSIÓN DEL VENCIMIENTO DE UN LOTE DE PRODUCTO FITOSANITARIO
Para los productos fitosanitarios vencidos o próximos a vencer, se podrá solicitar la extensión de la fecha de vencimiento de un lote dado de producto según el siguiente procedimiento:
1) Mediante la presentación de una solicitud, la firma registrante deberá identificar claramente el producto y el/los lote/s respectivo/s próximo/s a vencer o vencido/s.
2) SENASA procederá a realizar la toma de la/s muestra/s; O gestionar las mismas con la firma titular del producto.
3) La firma remitirá la/s muestra/s tomada/s a un laboratorio reconocido por SENASA para realizar los análisis pertinentes para comprobar que el lote del producto fitosanitario vencido o próximo a vencer se halla en condiciones para la extensión de su vida útil (cumple con la especificación).
4) Si corresponde extender la vida útil por un (1) año desde la fecha de vencimiento del lote, deberán obtenerse resultados conformes para la cuantificación de la/s sustancia/s activa/s de acuerdo con los límites establecidos en la Norma IRAM 12054.
5) Si corresponde extender la vida útil por dos (2) años desde la fecha de vencimiento del lote, deberán obtenerse resultados conformes para la cuantificación de la/s sustancia/s activa/s de acuerdo con los límites establecidos en la Norma IRAM 12054, y para el ensayo de estabilidad acelerada en almacenamiento.
6) La firma remitirá a SENASA el informe de los análisis efectuados junto con una nota, especificando: a. Marca comercial del producto b. Número de registro c. Lote d. Cantidad de unidades e. Tipo de presentación
7) En caso de poder extenderse la vida útil del lote en cuestión, el registrante deberá modificar la fecha de vencimiento (el resto de la información deberá mantenerse). Esto implica ubicar el nuevo vencimiento en el envase primario (y en el empaque secundario, si es que el producto lleva su vencimiento en la caja).
8) La firma registrante informará al Sistema Nacional de Trazabilidad la nueva fecha de vencimiento para el lote en cuestión, si así correspondiera.
REQUISITOS Y PROCEDIMIENTOS PARA PRODUCTOS DE TRATO DIFERENCIADO (PTD).
Se considera Producto de Trato Diferenciado a todo aquel que por sus características físicas y químicas, toxicológicas, ecotoxicológicas, modo de uso u otra cuestión técnica, no es alcanzado por la totalidad de los requisitos, o por su naturaleza o estado de avance técnico al momento de la presentación de la solicitud de registro.
Las personas físicas o jurídicas que inician un procedimiento de registro, podrán solicitar el tratamiento diferenciado de un principio activo o producto formulado, si corresponde, mediante la presentación de la información y documentación que fundamente dicho trato (Waiver), como anexo a cada tipo de categoría solicitada, a ser:
SUSTANCIAS ACTIVAS GRADO TÉCNICO NUEVAS.
SUSTANCIAS ACTIVAS GRADO TÉCNICO EQUIVALENTES.
PRODUCTOS FORMULADOS EN BASE A SUSTANCIAS ACTIVAS GRADO TÉCNICO NUEVAS O EQUIVALENTES.
COADYUVANTES DE APLICACIÓN, SEMIOQUÍMICOS Y FORMULADOS MISCELÁNEOS.
AUTORIZACIÓN DE PREDIOS EXPERIMENTALES Y DE USO EXPERIMENTAL DE SUSTANCIAS ACTIVAS SINTÉTICAS.
PRODUCTOS FORMULADOS EN BASE A SUSTANCIAS ACTIVAS BIOQUÍMICAS. PRODUCTOS FORMULADOS MICROBIANOS DESTINADOS A LA PROTECCIÓN VEGETAL
FISCALIZACIÓN
Fiscalización post autorización de la sustancia activa grado técnico nueva o equivalente o del producto formulado. El SENASA podrá efectuar fiscalizaciones post autorización de la sustancia activa grado técnico o del producto formulado mediante análisis de muestras del producto autorizado, estando el costo de envío de muestras y análisis a cargo del titular del registro o del importador. En caso de que la fiscalización demuestre inconsistencias entre la información declarada y los análisis efectuados sobre las muestras oficiales, el mencionado Servicio Nacional dará de baja la autorización de conformidad con los procedimientos establecidos en la normativa vigente, sin perjuicio de las sanciones que pudieran corresponder de acuerdo con lo dispuesto en el Capítulo V de la Ley N° 27.233 y su Decreto Reglamentario N° DECTO-2019-776-APN-PTE del 19 de noviembre de 2019, y la eventual aplicación de las medidas preventivas pertinentes.
Fiscalización documental post autorización de la sustancia activa grado técnico y/o del producto formulado. El SENASA podrá efectuar fiscalizaciones documentales post autorización de la sustancia activa grado técnico y/o del producto formulado. En caso de que la fiscalización demuestre inconsistencias entre la información declarada y la presentada al solicitar la autorización de la sustancia activa o del formulado para la planta en cuestión, el citado Servicio Nacional dependiendo del grado de inconsistencia dará de baja la autorización de conformidad con los procedimientos establecidos en la normativa vigente o solicitará la subsanación correspondiente, sin perjuicio de las sanciones que pudieran corresponder de conformidad con lo establecido en el Capítulo V de la mentada Ley N° 27.233 y su referido decreto reglamentario.
Las muestras oficiales de fiscalización y control serán remitidas a laboratorios que cuenten, al menos, con certificación de Buenas Prácticas de Laboratorio (BPL), es decir, que cumplan con las mismas exigencias de laboratorios que realicen estudios y análisis con objeto de evaluación para solicitud de registro.
En caso de detectarse alguna irregularidad relacionada con la calidad del producto o con la documentación declarada en productos con certificaciones de registro emitidas por las autoridades competentes de los países o grupos de países individualizados en el Anexo IV (IF-2025-68648006-APN-DNPV#SENASA), la autoridad competente podrá adoptará serán las siguientes acciones:
a) Suspender preventivamente el registro.
b) Dependiendo de la gravedad de lo hallado, se podrá solicitar la subsanación de la documentación presentada (bajo régimen de caducidad y pausado) y en caso de no ser posible la subsanación se procederá a la cancelación del registro.
c) dar de baja en el fitosanitarios en el VADEMECUM de acuerdo con la evaluación de las subsanaciones o cancelación.
d) De reiterarse TRES (3) irregularidades en las DDJJ del mismo o de distintos productos de una determinada firma registrantes, la Clave Única de Identificación Tributaria (C.U.I.T.) de la firma involucrada ingresará al régimen de vigilancia intensiva de monitoreo durante un año y dependiendo de la gravedad del incumplimiento se procederá a restringir por SEIS (6) meses la posibilidad de realizar cualquier actividad vinculada al comercio de fitosanitarios,
e) Estas medidas son tomadas sin perjuicio de aquellas otras medidas legales que pudieran adoptarse de acuerdo con la gravedad o el tipo de falta y las competencias intervinientes que correspondan.
IF-2025-121541245-APN-DNPV#SENASA

LISTADO DE PAÍSES/GRUPO DE PAÍSES CON CONVERGENCIA NORMATIVA
A continuación, se establece el listado de países/grupo de países con convergencia normativa con la REPÚBLICA ARGENTINA:
MANCOMUNIDAD DE AUSTRALIA,
CANADÁ,
CONFEDERACIÓN SUIZA,
UNIÓN EUROPEA,
ESTADOS UNIDOS DE AMÉRICA,
ESTADO DEL JAPÓN,
ISLANDIA,
PRINCIPADO DE LIECHTENSTEIN,
REINO DE NORUEGA,
NUEVA ZELANDA,
REINO UNIDO DE GRAN BRETAÑA E IRLANDA DEL NORTE,
REPÚBLICA FEDERATIVA DE BRASIL.

ANEXO IV
(Anexo sustituido por art. 9° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
(Anexo sustituido por art. 9° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
LISTADO DE PAÍSES/GRUPO DE PAÍSES CON CONVERGENCIA NORMATIVA
A continuación, se establece el listado de países/grupo de países con convergencia normativa con la REPÚBLICA ARGENTINA:
MANCOMUNIDAD DE AUSTRALIA,
CANADÁ,
CONFEDERACIÓN SUIZA,
UNIÓN EUROPEA,
ESTADOS UNIDOS DE AMÉRICA,
ESTADO DEL JAPÓN,
ISLANDIA,
PRINCIPADO DE LIECHTENSTEIN,
REINO DE NORUEGA,
NUEVA ZELANDA,
REINO UNIDO DE GRAN BRETAÑA E IRLANDA DEL NORTE,
REPÚBLICA FEDERATIVA DE BRASIL.
IF-2025-121541330-APN-DNPV#SENASA
ANEXO V
AVISO DE IMPORTACIÓN DE PRODUCTOS FITOSANITARIOS
SUSTANCIAS ACTIVAS GRADO TÉCNICO NUEVAS, EQUIVALENTES Y/O PRODUCTOS FORMULADOS.
Cuando los productos fitosanitarios cuenten con la aprobación de uso agrícola, por las Autoridades Competentes del listado de países o grupos de países que se detallan en el Anexo IV de la presente norma, deberán cumplir con lo prescripto en su Artículo 7°, según el procedimiento que a continuación se detalla:
I. INFORMACIÓN ADMINISTRATIVA
1) Formulario con carácter de declaración jurada, informando el Grado Técnico Nuevo o Equivalente y/o productos formulados a través de la plataforma SIG-Trámites del SERVICIO NACIONAL DE SANIDAD Y CALIDAD AGROALIMENTARIA (SENASA) o la que en el futuro la reemplace, firmada por el Apoderado o Representante Legal y el Responsable Técnico con incumbencia profesional en la materia.
DATOS DEL SOLICITANTE:
Nombre y Apellido;
Razón Social;
Dirección;
Teléfono;
Correo Electrónico;
DATOS DEL PRODUCTO A IMPORTAR:
Nombre químico;
Número de código experimental;
Grupo químico;
Concentración de Sustancia Activa;
Características/Estado Físico;
Establecimiento productor,
País de origen de la sustancia;
2) Presentar el documento oficial de registro y/o comercialización emitido por la Autoridad Competente que integre el listado de países mencionado en el Anexo IV de la presente norma, donde conste la denominación de la sustancia activa grado técnico, la pureza mínima expresada en PORCENTAJE PESO EN PESO (% P/P), el nombre y el domicilio del establecimiento sintetizador.
3) Hoja de Datos de Seguridad, de acuerdo con la versión del Manual de las Naciones Unidas del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA), adoptada por la Autoridad Competente del Registro Nacional.
En el caso de que los productos NO se comercialicen en la REPÚBLICA ARGENTINA, se deberá declarar además de lo solicitado en los Puntos 1, 2, y 3, lo siguiente:
- el límite máximo de residuos (LMR) establecido en el país en el que el producto se encuentra registrado, indicando las condiciones bajo las cuales dicho límite fue determinado;
- el LMR establecido en el país en el que el producto se encuentra registrado para el o los cultivos a tratar, y el límite de cuantificación analítica (LOQ);
- el cultivo objetivo al que está destinado;
- organismo nocivo o plaga a ser controlada,
- la dosis de aplicación recomendada,
- período de carencia para cada cultivo,
- Registro Nacional Sanitario de Productores Agropecuarios (RENSPA) donde será utilizado.
La finalidad de esta declaración jurada es demostrar que el producto fitosanitario no representa un riesgo para la salud humana, animal ni para el ambiente.
Alternativamente, el solicitante podrá optar por presentar los resultados de ensayos de eficacia agronómica y LMR realizados en la REPÚBLICA ARGENTINA, a fin de acreditar que el producto no incurre en riesgos para la salud humana, animal ni ambiental en las condiciones locales de uso.
II. INFORMACIÓN Y DOCUMENTACIÓN CONFIDENCIAL.
Información confidencial:
- Declaración de pureza e impurezas para sustancias activas.
- Declaración de composición en los formulados.
La información y documentación confidencial será reservada al sólo efecto de la fiscalización, y para el caso de sustancias activas nuevas, como referencia de sustancias activas equivalentes.
IF-2025-68648196-APN-DNPV#SENASA
ANEXO VI
(Anexo sustituido por art. 9° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
(Anexo sustituido por art. 9° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
AVISO DE IMPORTACIÓN DE MUESTRAS DE PRODUCTOS FITOSANITARIOS EN ETAPAS TEMPRANAS DE DESARROLLO
Para cada sustancia o producto de la que se desee importar una muestra, se deberá presentar un aviso de importación con carácter de declaración jurada, la cual será automática, a través de la Plataforma SIG-Trámites o la que en el futuro la reemplace, que contenga la siguiente información:
Datos del solicitante:
1. Nombre;
2. Dirección;
3. Teléfono;
4. Correo Electrónico;
5. Responsable legal
1. Datos del producto a importar:
1. Nombre químico;
2. Número de código experimental;
3. Grupo químico;
4. Concentración de Sustancia activa;
5. Características/estado físico;
6. Establecimiento productor, país de origen de la sustancia;
7. Destino dentro del país y/o localidad en que se realizará la prueba (Predio de experimentación);
8. Cantidad de sustancia, en unidades y/o kilogramos y/o litros;
9. Justificación de la cantidad de muestra necesaria para las pruebas en ejecución y/o en proyecto,
10. Hoja de Datos de Seguridad, de acuerdo a la versión del manual de las Naciones de las Naciones Unidas del “Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos”, adoptada por la Autoridad Competente del Registro Nacional de Terapéutica Vegetal.
Las muestras en etapa de desarrollo no podrán ser comercializadas, dado que estas están destinadas exclusivamente a uso experimental. Su ingreso al país se realizará bajo la entera responsabilidad de quien las introduzca, debiendo garantizar el cumplimiento de las normativas vigentes y las condiciones establecidas para su manipulación y uso.
Las muestras deberán tener en su identificación adherida al envase la frase: 'MUESTRA SIN VALOR COMERCIAL, PROHIBIDA SU VENTA'.
ANEXO VII
REEVALUACIÓN DE PRODUCTOS FITOSANITARIOS REGISTRADOS
Después de la aprobación de un componente activo o del registro de un producto fitosanitario, puede haberse generado nueva información científica que sugiera la existencia de riesgos para la salud humana, la seguridad animal o el medio ambiente previamente desconocidos, así como también en el comercio de productos agrícolas o incluso que surgieran datos e información asociados a problemas o disminución de la eficacia agronómica de un producto fitosanitario.
El proceso de reevaluación o revisión de productos fitosanitarios registrados tiene como propósito que, mediante la herramienta de análisis de riesgo, la Autoridad Competente determine si en función de nuevos antecedentes técnicos y científicos respecto a la caracterización de peligros, evaluación de la exposición, cambios en la eficacia de los productos autorizados debido a cambios tecnológicos o aparición de resistencia de plagas, entre otras cuestiones, resulta procedente iniciar los procedimientos para modificar prácticas agrícolas, períodos de carencia, límites máximos de residuos, vigencia de las autorizaciones, realizar reclasificaciones, establecer restricciones parciales o totales de un producto fitosanitario o cualquier otra medida preventiva o correctiva cuando los usos autorizados vigentes de ese producto puedan causar efectos adversos, en las condiciones locales de uso, en niveles inaceptables tanto para la salud como para el ambiente o pueda verse afectada la eficacia agronómica en las condiciones locales de dicho producto o afecte el comercio de los productos agrícolas o alimentos que se comercialicen.
De este modo, la revisión de registros tiene como objetivo garantizar que, a medida que evoluciona la capacidad de evaluar riesgos y cambian las caracterizaciones de las sustancias y las prácticas, todos los productos fitosanitarios registrados sigan cumpliendo con el estándar legal de ausencia de efectos adversos inaceptables a la salud y el ambiente.
Sin perjuicio de la aplicación del proceso de reevaluación o revisión sobre productos fitosanitarios por las razones indicadas, la Autoridad Competente puede tomar las medidas preventivas o correctivas que correspondan sin que medie revisión, si las razones que ameritan tales medidas se fundamentan en cuestiones comerciales o situaciones de emergencia que requieran de ellas, pudiendo abstraerse de la reevaluación o revisión.
MOTIVOS DE INICIO DE REEVALUACIÓN O REVISIÓN
La Autoridad Nacional Competente del Registro Nacional podrá realizar un plan de priorización plurianual para la reevaluación de usos locales de sustancias activas relacionadas con posibles cambios en el perfil de riesgo en la i) Exposición alimentaria; ii) Exposición laboral; iii) Exposición ambiental; y iv) Eficacia agronómica/resistencia de plagas, así como también debido al surgimiento de nueva información disponible sobre los perfiles toxicológicos, ecotoxicológicos y el destino ambiental de las sustancias activas.
PROCEDIMIENTO
El proceso formal de reevaluación o revisión de productos fitosanitarios registrados se inicia cuando la Autoridad Competente envía una notificación a los registrantes, o publica oficialmente una decisión técnica debidamente justificada, informando su decisión de someter las autorizaciones de uso aprobadas, y/o registros, a revisión.
El cumplimiento de este aviso es un requisito legal para los titulares que deseen mantener las autorización de uso vigente, y se aplicarán sanciones por no proporcionar la información requerida dentro del plazo especificado en el plan de trabajo propuesto. El plan de trabajo para la reconsideración será discutido con los titulares de registro y se establecerán hitos clave y el plazo normativo dentro del cual se debe tomar una decisión sobre las condiciones de autorización de usos en evaluación.
Además de los datos presentados por los titulares de registro, la autoridad competente podrá considerar todos los datos publicados e informes de evaluación relevantes recabados por otras agencias regulatorias.
Los estudios requeridos que surgieran del plan de trabajo serán responsabilidad compartida por todas las empresas afectadas y/o empresas interesadas en participar en conjunto, siendo la distribución de tareas y costos responsabilidad de las propias empresas, debiendo estas proceder a su distribución en base a los costos efectivos de los estudios realizados y las participaciones respectivas en el mercado local.
DECISIÓN DE REEVALUACIÓN O REVISIÓN
Todas las empresas afectadas deben aceptar las decisiones de reevaluación o revisión de la Autoridad Competente, las cuales pueden estar fundadas en los motivos enumerados a continuación:
- Adecuación de límites máximos de residuos y tiempos de carencia.
- Cancelación de usos.
- Adecuación de dosis y/o momentos de aplicación.
- Modificación del espectro de control de plagas informado previamente.
- Restricción de formas de aplicación.
- Reclasificación de peligro/riesgo.
- Cancelación de determinadas formulaciones.
- Cancelación de registros.
- Toda otra modificación, restricción o cancelación, no contemplada expresamente.
MEDIDAS TRANSITORIAS SOBRE PRODUCTOS EN REEVALUACIÓN O REVISIÓN
Pueden recibirse solicitudes de registro de sustancias activas y sus productos formulados puestos en reevaluación o revisión debido a nuevos antecedentes técnicos y científicos respecto a la caracterización de peligros toxicológicos o ecotoxicológicos, a cambios en la eficacia o aparición o existencia confirmada de resistencia de plagas.
No se aceptarán nuevos usos o modificaciones de recomendaciones de uso de las sustancias en revisión mientras se encuentre activa la reevaluación y hasta tanto la Autoridad Competente se expida sobre la decisión a adoptarse como resultado de la revisión.
Las personas humanas o jurídicas que presenten solicitudes de registro de productos fitosanitarios que se encuentren en reevaluación deben declarar su conocimiento del plan de trabajo comunicado y de los hitos clave y los plazos establecidos, así como también el compromiso de su cumplimiento.
IF-2025-68651146-APN-DNPV#SENASA
ANEXO VIII
(Anexo sustituido por art. 9° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
(Anexo sustituido por art. 9° de la Resolución N° 843/2025 del Servicio Nacional de Sanidad y Calidad Agroalimentaria B.O. 4/11/2025. Vigencia: a partir del día de su publicación en el Boletín Oficial.)
CLASIFICACIÓN, COMUNICACIÓN DE LOS PELIGROS PARA LA SALUD, Y PROTOCOLO DE ENSAYOS DE EFICACIA AGRONÓMICA, FITOTOXICIDAD Y RESIDUOS
PROTOCOLO DE ENSAYOS DE EFICACIA AGRONÓMICA Y FITOTOXICIDAD
PROPÓSITO Y ALCANCES:
El objetivo de los ensayos de eficacia agronómica es proporcionar resultados comparables y confiables sobre la eficacia de un producto fitosanitario a registrar o de su recomendación de uso, considerando las condiciones climáticas y agronómicas locales, de acuerdo con las Buenas Prácticas Agrícolas (BPA) propuestas en el marbete del producto fitosanitario. Los resultados de los ensayos de eficacia y fitotoxicidad son la base para el establecimiento de la Buena Práctica Agrícola Crítica que se utilizará para la definición del LMR para el uso propuesto.
REQUISITOS DE PRESENTACIÓN:
La Autoridad Competente deberá solicitar ensayos de eficacia agronómica para aquellos productos fitosanitarios registrados o en vías de registro, cuando deba reunir evidencias significativas para:
- Cuantificar la eficacia agronómica de un producto formulado a registrar con base en sustancias activas grado técnico nuevas.
- Cuantificar la eficacia agronómica de un producto formulado registrado con base en sustancias activas grado técnico equivalentes, en un nuevo cultivo.
- Cuantificar la eficacia agronómica de un producto formulado registrado con base en sustancias activas grado técnico equivalentes, para el control de una nueva plaga.
- Cuantificar la eficacia agronómica de un producto formulado registrado con base en sustancias activas grado técnico equivalentes, ante un cambio de dosis o momento de aplicación.
- Cuantificar la eficacia agronómica de una nueva combinación de principios activos o el cambio en la proporción de sustancias activas grado técnico equivalentes, en un producto formulado en mezcla.
PROCESO DE VERIFICACIÓN:
La empresa registrante debe notificar semestralmente ante la Autoridad Competente, la planificación semestral de ensayos de eficacia agronómica. En dicha planificación debe indicar los sitios donde se realizarán los ensayos de eficacia agronómica, la fecha estimada de inicio y de las evaluaciones postaplicación y los datos de contacto del ensayista responsable, a fin de que la autoridad regulatoria pueda realizar el seguimiento y solicitar información adicional cuando lo considere necesario con fines de verificación.
Los ensayos podrán ser constatados o verificados por la Autoridad Competente en aquellos puntos que considere críticos del proceso del ensayo de eficacia agronómica (a saber: dosificación, aplicación, monitoreo u otro), por lo que cada una de estas actividades deben estar debidamente registradas en los medios físicos y/o digitales que el ensayista responsable considere adecuados.
REQUISITOS MÍNIMOS DE PLANIFICACIÓN DE ENSAYOS LOCALES:
Los ensayos de eficacia agronómica deberán llevarse a cabo donde el cultivo en estudio es realizado comercialmente, con niveles apropiados de infecciones o infestaciones de plagas que permitan verificar la eficacia de los tratamientos evaluados. Deben reflejar las recomendaciones de uso que se deberán incorporar en la etiqueta del producto.
Al menos SEIS (6) ensayos de eficacia agronómica deberán llevarse a cabo en diferentes zonas agroecológicas de la REPÚBLICA ARGENTINA, representativas de la combinación plaga-cultivo para la que solicita la ampliación de uso. Los ensayos deberán realizarse durante al menos DOS (2) campañas agrícolas.
En casos excepcionales, se podrán aceptar protocolos de ensayos de eficacia y fitotoxicidad en una campaña agrícola, previa justificación técnica a la Autoridad Competente que motive el trato diferenciado. En tales casos, los lotes experimentales ubicados en una misma región agroecológica, deben estar distanciados al menos VEINTE KILÓMETROS (20 km) entre sí.
Para cultivos considerados menores en la REPÚBLICA ARGENTINA, se requerirán al menos DOS (2) ensayos locales.
RESIDUOS EN PRODUCTOS TRATADOS
El plan de ensayos de estudios de residuos locales puede desarrollarse considerando tres categorías de cultivos:
- Cultivos mayores, representativos en Argentina.
- Cultivos menores.
- Cultivos mayores, NO representativos en Argentina.
ESTABLECIMIENTO DE RESIDUO POR ESPECIE VEGETAL, EN CULTIVOS MAYORES REPRESENTATIVOS EN ARGENTINA:
Se requerirán al menos SEIS (6) ensayos de residuos, de los cuales, al menos CUATRO (4) deberán ser locales, realizados en áreas agroecológicas diferentes y representativas de cada cultivo, en la misma temporada o en temporadas diferentes (ver Tabla Agrupación de Cultivos, mayores representativos y de cultivos menores para la REPÚBLICA ARGENTINA). Optativamente, serán aceptados hasta DOS (2) estudios de residuos realizados bajo Buenas Prácticas de Laboratorio (BPL) conducidos en otros países, siempre que la recomendación de uso del ensayo sea comparable con la práctica agrícola crítica a registrar.
ESTABLECIMIENTO DE RESIDUO POR GRUPOS DE CULTIVO, SOBRE LA BASE DE CULTIVOS MAYORES REPRESENTATIVOS EN ARGENTINA:
Se requerirán al menos OCHO (8) ensayos de residuos locales, en especies representativas pertenecientes a los grupos de cultivos definidos en el presente Anexo (ver Tabla Agrupación de Cultivos, mayores representativos y de cultivos menores para la REPÚBLICA ARGENTINA), en áreas agroecológicas diferentes y representativas de cada cultivo, en la misma temporada o en temporadas diferentes. Optativamente, serán aceptados hasta el VEINTICINCO POR CIENTO (25 %) del número total de estudios de residuos BPL, que hayan sido conducidos en otros países, siempre que la recomendación de uso del ensayo sea comparable con la práctica agrícola crítica a registrar.
ESTABLECIMIENTO DE RESIDUO POR GRUPOS DE CULTIVO Y POR ESPECIE VEGETAL EN CULTIVOS MAYORES NO REPRESENTATIVOS EN ARGENTINA:
Se requerirán al menos SEIS (6) ensayos de residuos BPL para fijación de tolerancia por grupos, y CUATRO (4) para fijación de tolerancias por especie mayor NO representativa de Argentina, los cuales pueden haber sido conducidos en otros países, siempre que la recomendación de uso del ensayo sea comparable con la práctica agrícola crítica a registrar (ver Tabla Agrupación de Cultivos, mayores NO representativos y de cultivos menores para la REPÚBLICA ARGENTINA).
ESTABLECIMIENTO DE RESIDUO POR ESPECIE VEGETAL, EN CULTIVOS MENORES EN ARGENTINA:
Se requerirán al menos DOS (2) ensayos de residuos BPL conducidos en Argentina o en otros países, siempre que la recomendación de uso del ensayo sea comparable con la práctica agrícola crítica a registrar (ver Tabla Agrupación de Cultivos, mayores representativos y de cultivos menores para la REPÚBLICA ARGENTINA y Tabla Agrupación de Cultivos, mayores NO representativos y de cultivos menores para la REPÚBLICA ARGENTINA);
Se considerarán excepciones a estos requisitos:
- Establecimiento de residuos para uso en post-cosecha:
Se requerirán al menos CUATRO (4) ensayos de residuos por especie vegetal o en cultivos representativos del correspondiente grupo de cultivos según lo establecido en las Tabla Agrupación de Cultivos, mayores representativos y de cultivos menores para la REPÚBLICA ARGENTINA y Tabla Agrupación de Cultivos, mayores NO representativos y de cultivos menores para la REPÚBLICA ARGENTINA. Estos ensayos pueden ser desarrollados localmente o conducidos en otros países, en este último caso, siempre que la recomendación de uso del ensayo sea comparable con la práctica agrícola crítica a registrar en Argentina.
- Aplicación en pre-siembra o en preemergencia del cultivo y aplicación de herbicidas entre las líneas de cultivos perennes en producción, aplicación en dormancia de cultivos perennes o cuando se cuente con ensayos presentados ante Autoridades regulatorias, hechos bajo BPL y que cumplan con la práctica crítica (BPA) a registrar, que demuestren que los residuos se encontrarán por debajo del límite de cuantificación:
Se requerirán DOS (2) ensayos de campo en regiones agroecológicas diferentes y representativas del cultivo, en la misma estación o en temporadas diferentes en el mismo lugar, si el resultado de la los valores de residuos son menores o iguales a un Límite de Quantificación (LOQ) de CERO COMA CERO UNA PARTES POR MILLÓN (0,01 ppm).
Sin embargo, si el resultado del estudio de residuo es mayor que el valor del LOQ, en al menos UNA (1) de las pruebas, se deberán realizar ensayos adicionales que cumplan con los criterios detallados en los puntos “Establecimiento de residuo por especie vegetal, en cultivos mayores representativos en Argentina” y “Establecimiento de residuo por grupos de cultivo, sobre la base de cultivos mayores representativos en Argentina”.
RESTRICCIONES DE USO DE MATERIAL TRATADO:
Los productos agrícolas y los restos de cultivo, provenientes de las áreas tratadas con los productos formulados a evaluar, no podrán ser utilizados para alimentación humana o animal. Cuando se trate de un ensayo de experimentación de productos utilizados en forma de trampas o cebos, que por su forma de uso no entren en contacto con el cultivo, podrá permitirse el consumo de los productos agrícolas para fines de alimentación humana o animal.
ANÁLISIS DE RESULTADOS:
Se deben presentar los resultados obtenidos tras la aplicación de diversas técnicas estadísticas a los datos.
En el texto del informe deben figurar solo los datos estadísticos principales, la información sobre la media y los errores estándar de las variables cuantificadas y comparadas entre los tratamientos evaluados.
AMPLIACIONES DE USO DE OFICIO PARA CULTIVOS MENORES O USOS MENORES
La Autoridad Competente puede, sobre la base de información suficiente y científicamente válida desarrollada en el país o en el exterior, establecer prácticas agrícolas y límites máximos de residuos de oficio en forma temporal o definitiva, dependiendo de la calidad de la información que disponga.
A tal fin, puede conformar comisiones o grupos de trabajo con la participación del INSTITUTO NACIONAL DE TECNOLOGÍA AGROPECUARIA (INTA), Universidades, expertos y especialistas en la materia, representantes de las áreas competentes en materia de productos fitosanitarios de los gobiernos provinciales, representantes de productores, organizaciones o instituciones de investigación y otras que con incumbencia en la materia colaboren en el desarrollo y autorización de usos en cultivos menores o usos menores con sustento científico.
CONCLUSIONES:
Pueden brindarse análisis comparativos de los resultados de esta investigación con los de otros autores, para evidenciar la contribución neta realizada por los ensayos, la corroboración de otros estudios, las discrepancias con ellos, etcétera. Cualquier información que respalde específicamente las declaraciones de etiqueta debe ser discutida, incluidos los efectos secundarios, si los hubiera.
Todos los ensayos que realicen los interesados y la información que surja de ellos sobre composición y procesos de elaboración serán reservados. Su vista queda reservada a personal y auxiliares de la Autoridad Competente afectada al proceso aprobatorio del producto, a los responsables técnicos designados y a las personas fehacientemente autorizadas por la firma que solicita la inscripción.
NOTA ACLARATORIA
Las tablas detalladas a continuación son a modo de guía conceptual, complementa, pero no reemplaza los criterios definidos en la Versión ST/SG/AC.10/30/Rev.9 del Manual de las Naciones Unidas del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA).
LAS TABLAS NO SE AGREGAN YA QUE NO TUVIERON COMENTARIOS.
NOTA ACLARATORIA
Las tablas detalladas a continuación son a modo de guía conceptual, complementa, pero no reemplaza los criterios definidos en la Versión ST/SG/AC.10/30/Rev.9 del Manual de las Naciones Unidas del Sistema Globalmente Armonizado de Clasificación y Etiquetado de Productos Químicos (SGA).
Fuente: FAO and WHO. 2022. Guidance on good labelling practice for pesticides (Second revision)
- International Code of Conduct on Pesticide Management. Rome.
Criterios que definen las categorías de peligro de toxicidad aguda
Notas a la tabla:
a) La estimación de la toxicidad aguda (ETA) para la clasificación de una sustancia se deducirá a partir de la DL50/CL50, cuando se conozca;
b) Los valores de toxicidad aguda se expresan en valores (aproximados) de la DL50 (ingestión, absorción cutánea) o CL50 (inhalación) o en estimaciones de la toxicidad aguda (ETA).
c) Los valores de corte/límites de concentración para la toxicidad por inhalación que figuran en la tabla se basan en una exposición de CUATRO (4) horas. Para convertir los datos de modo que respondan a una exposición de UNA (1) hora, hay que dividirlos por DOS (2) para gases y vapores y por CUATRO (4) para polvos y nieblas;
d) Algunas reglamentaciones utilizan la concentración de vapor saturado como un elemento adicional para protecciones específicas de salud y seguridad (por ejemplo, la Reglamentación Modelo de las Naciones Unidas);
e) En algunas sustancias, la atmósfera del ensayo no será solo un vapor sino que consistirá en una mezcla de fases líquidas y gaseosas. En otras sustancias, esa atmósfera podrá consistir en un vapor próximo al estado gaseoso. En estos últimos casos, la clasificación [en partes por millón de volumen (ppmV)] será la siguiente: Categoría 1, CIEN PARTES POR MILLÓN DE VOLUMEN (100 ppmV); Categoría 2, QUINIENTAS PARTES POR MILLÓN DE VOLUMEN (500 ppmV); Categoría 3, DOS MIL QUINIENTAS PARTES POR MILLÓN DE VOLUMEN (2.500 ppmV); Categoría 4, VEINTE MIL PARTES POR MILLÓN DE VOLUMEN (20.000 ppmV). Los términos “polvo”, “niebla” y “vapor” se definen como sigue:
- Polvo: partículas de una sustancia o de una mezcla en suspensión en un gas (en el aire por lo general)
- Niebla: gotas líquidas de una sustancia o de una mezcla en suspensión en un gas (en el aire por lo general)
- Vapor: forma gaseosa de una sustancia o de una mezcla liberada a partir de su estado líquido o sólido.
El polvo se forma generalmente por un proceso mecánico. Las nieblas se forman generalmente por condensación de vapores supersaturados o por el fraccionamiento físico de líquidos. El tamaño de polvos y nieblas oscila generalmente entre valores que van desde menos de UNO (1) hasta alrededor de CIEN MICRÓMETROS (100 |im);
f) Los valores para polvos y nieblas deberían revisarse para adaptarse a futuros cambios de las directrices de la Organización para la Cooperación y el Desarrollo Económico (OCDE) con respecto a las limitaciones técnicas en la generación, mantenimiento y medición de las concentraciones de polvos y nieblas en forma respirable;
g) Los criterios de la Categoría 5 se proponen identificar las sustancias que presenten un peligro relativamente bajo de toxicidad aguda, pero que en determinadas circunstancias puedan suponer un peligro para poblaciones vulnerables. La DL50 de esas sustancias se sitúa en el rango de DOS MIL A CINCO MIL MILIGRAMOS POR KILOGRAMO (2.000-5.000 mg/kg) de peso corporal y en dosis equivalentes para la inhalación. Los criterios específicos de la Categoría 5 son:
i. La sustancia se clasifica en esta categoría, si ya se dispone de información fidedigna que indique que la DL50 (CL50) corresponde al rango de valores de la Categoría 5 o cuando otros estudios con animales o sobre los efectos tóxicos agudos en seres humanos constituyen un motivo de preocupación para la salud humana;
ii. La sustancia se clasificará en esta categoría, mediante extrapolación, estimación o medición de datos, cuando no esté justificada su asignación a una categoría de mayor peligro; y:
- Se disponga de información fidedigna sobre la existencia de efectos tóxicos significativos en los seres humanos; o
- Se observe mortalidad en los ensayos sobre exposición por vía oral o cutánea o por inhalación, con valores hasta de la Categoría 4; o
- Cuando la opinión de los expertos confirme la aparición de síntomas clínicos de toxicidad significativos (excepto diarrea, piloerección o aspecto descuidado) en ensayos realizados con valores hasta de la Categoría 4; o
- Cuando tal opinión confirme la existencia de información fidedigna sobre efectos agudos potencialmente significativos procedentes de otros estudios con animales.
Tabla: Categorías y subcategorías de corrosión dermal.
Tabla: Elementos de corrosión/irritación dermal que deben figurar en una etiqueta.
Tabla: Categoría correspondiente a lesiones oculares graves/efectos irreversibles en los ojos.
Tabla: Categorías de efectos reversibles en los ojos.
Tabla: Elementos que deben figurar en la etiqueta para lesiones oculares graves/irritación ocular.
Tabla: Categoría y subcategorías de peligro para los sensibilizantes respiratorios.
Tabla: Categoría y subcategorías de peligro para los sensibilizantes dermales.
Tabla: Elementos que deben figurar en las etiquetas para sensibilizantes respiratorios y cutáneos.
Tabla: Categorías para las sustancias peligrosas para el medio ambiente acuático. Peligro a corto plazo (agudo).
Tabla: Elementos que deben figurar en las etiquetas de peligro para sustancias y mezclas peligrosas para el medio ambiente acuático.
PELIGRO A CORTO PLAZO (AGUDO) PARA EL MEDIO AMBIENTE ACUÁTICO
PELIGRO A LARGO PLAZO (CRÓNICO) PARA EL MEDIO AMBIENTE ACUÁTICO
Tabla: Categorías para las sustancias peligrosas para aves y elementos que deben figurar en la etiqueta.
Tabla: Categorías para las sustancias peligrosas para abejas y elementos que deben figurar en la etiqueta.
Tabla: Pictogramas de precaución para reducir riesgos al manipular, aplicar o almacenar un producto fitosanitario.
Tabla: Agrupación de Cultivos, mayores representativos y de cultivos menores para la REPÚBLICA ARGENTINA.
Tabla: Agrupación de Cultivos, mayores NO representativos y de cultivos menores para la REPÚBLICA ARGENTINA.
IF-2025-121331444-APN-DNPV#SENASA